Chapter 6:
Pathogenesis of TBEV-diseases
Anna K Överby, Saravanan Thangamani
Key points
- In this chapter we describe the pathogenesis of tick-borne encephalitis virus (TBEV).
- To cause infection, TBEV needs to cross three different barriers; the physical, the innate and adaptive and the blood-brain barrier.
- TBEV transmission at the skin interface is pro-inflammatory with a marked increase in immune cell infiltrates at the tick-feeding foci.
- The trigger of innate immune and adaptive immune responses, by TBEV is necessary to clear the infection.
- TBEV employs different strategies to evade the innate immune response.
- Both different animal models and reverse genetics will help us understand TBEV pathogenesis.
Transmission and entry:
Tick vectors and tick-host interface
The Ixodes ricinus tick serves as the primary carrier of TBEV-Eu in nature, while the Ixodes persulcatus tick is the primary vector for TBEV-Sib and TBEV-FE.1 I. ricinus is widely spread across Europe, reaching into Turkey and northern Iran, whereas I. persulcatus is found in the Urals, Siberia, Far-Eastern Russia, as well as parts of China and Japan.2,3 A zone of sympatry exists in the northern Baltics, western Finland, and northwestern Russia, where the habitats of I. ricinus and I. persulcatus overlap, leading to the presence of multiple TBEV subtypes.3-5 TBEV is maintained within natural transmission cycles involving ixodid ticks and wild-living mammalian hosts. Infected ticks are presumed to remain infected throughout their life cycle.2 While transovarial transmission of TBEV from an infected female tick to the egg mass is possible, this mode of infection is not entirely efficient in sustaining TBEV within the natural tick population.6
The transmission of tick-borne encephalitis virus (TBEV) from an infected tick to a host involves a complex interplay between the tick’s feeding process and the immunomodulatory properties of its saliva. This process begins shortly after the tick attaches itself to the host. TBEV is transmitted to the vertebrate host along with the tick’s saliva as early as one hour after the tick attaches7 and POWV is transmitted as fast as 15 minutes after attachment.8 Tick feeding is a sophisticated process, and successful feeding is facilitated by various components present in the tick’s saliva, which possess immunomodulatory properties. Notably, tick salivary factors not only aid in blood feeding but also modulate the host environment, thereby promoting the transmission and establishment of TBEV.9
Seminal studies conducted by Labuda et al. (1993) demonstrated the significance of saliva-assisted transmission (SAT) of TBEV.10 They observed that when naïve guinea pigs were inoculated with a mixture of TBEV and salivary gland extract (SGE) obtained from partially fed uninfected female ticks of species like Ixodes ricinus, Dermacentor reticulatus, or Rhipicephalus appendiculatus, and subsequently, uninfected Rhipicephalus appendiculatus nymphs fed on these guinea pigs, there was an increased acquisition of the virus by ticks feeding on animals inoculated with the mixture of SGE and virus compared to those inoculated with the virus alone. This research underscores the crucial role of tick saliva in facilitating the transmission of TBEV and sheds light on the mechanisms involved in the transmission dynamics between ticks and hosts. Observations of pathogens being transmitted from infected ticks to uninfected ticks co-feeding on the same host have offered indirect evidence of what is known as “sequential acquisition of tick-borne pathogens,” as noted by Nuttall and Labuda in 2004.9 It is also referred to as co-feeding transmission. In natural environments, it’s common for infected ticks to co-feed alongside uninfected ticks on a single host. Labuda et al. conducted experiments where TBEV-infected I. ricinus ticks and uninfected ticks co-fed on naïve, natural host species. Intriguingly, they found that the highest numbers of TBEV-infected ticks originated from susceptible host species with very low levels of viremia, providing compelling evidence that non-viremic co-feeding transmission of TBEV is a primary mechanism for maintaining the virus in natural foci.11,12
Tick-host-virus interface during TBEV transmission:
Skin acts as the primary barrier against various forms of damage, including mechanical stress, environmental factors, and potential infections. It serves as the frontline defense between a tick and its host, making it the first point of contact for both TBEV and tick saliva during feeding. Throughout the feeding process, a tick’s mouthparts and saliva interact with the host’s blood and lymphatic vessels, as well as various cellular components such as fibroblasts, keratinocytes, Langerhans cells, dendritic cells, macrophages, mast cells, natural killer cells, T lymphocytes, and soluble mediators like cytokines, chemokines, complement proteins, and lectins.13 These cutaneous immune cells play a pivotal role in initiating the host’s immune response and inflammatory reactions against tick feeding and potential pathogen transmission.
The significance of skin infection in the transmission of TBEV is paramount. Skin acts as the primary interface where these viruses establish infection in the host.9 Labuda et al. thoroughly investigated the initial stages of TBEV replication within the skin of two natural host species: bank voles (Clethrionomys glareolus) and yellow-necked field mice (Apodemus flavicollis). Their experimental setup mirrored natural conditions, with infected and uninfected Ixodes ricinus ticks placed on specific areas of the host’s skin. Their findings revealed a correlation between TBEV detection in feeding ticks and the transmission dynamics from infected to uninfected ticks.14 Additionally, TBEV exhibited a preference for skin sites where ticks were actively feeding. To characterize TBEV-infected cells, Labuda et al. infested laboratory mice with TBEV-infected ticks and cultured skin explants from the infestation sites. They observed the migration of leukocytes from these explants, with viral antigens present in migrating Langerhans cells and neutrophils, indicating their role in viral dissemination.14 In vitro studies suggest that dendritic cell populations at the tick feeding site are among the early targets of TBEV infection. Recent research indicates that exposure of bone marrow-derived dendritic cells to tick saliva enhances TBEV replication, partly through activation of the pro-survival Akt pathway.15
These results underscore the importance of localized skin infection in the early transmission of the virus from infected ticks and its acquisition by uninfected co-feeding ticks.11,16 Immune cells infiltrating the skin during tick feeding act as carriers for virus transmission between co-feeding ticks, independent of systemic viremia.14 Langerhans cells, the primary dendritic cell population in the epidermis, likely play a crucial role in virus dissemination, as evidenced by their migration to draining lymph nodes in response to cutaneous infections with other arthropod-borne viruses.17 Thus, the presence of TBE viral antigen in emigrating Langerhans cells suggests their involvement in transporting TBEV to the lymphatic system, contributing to overall viral dissemination. The importance of virus-infected cells at the tick feeding site and their contribution to initial viral replication and dissemination was further supported by in vitro experiments where I. ricinus tick saliva was shown to modulate TBEV infection of dendritic cells. Specifically, when DCs were cultured with TBEV in the presence of I. ricinus saliva, the infection rate of the cells was enhanced and there was a decrease in virus-induced TNF- alpha and IL6 production.18
A study conducted by Thangamani et al. explored the immune response in the skin to TBEV infection. The study involved allowing TBEV-infected ticks to feed on mice, followed by biopsies of the bite sites at one and three hours post-attachment for RNAseq transcriptome and histochemical analysis. The analysis revealed upregulation of various cytokines (Ccl2, Ccl12, Cxcl1, Cxcl2, Cxcl5, IL6, and IL10) and receptors (CCR1, CCR5, and Sell) after just one hour of TBEV-infected tick feeding, indicating an early activation of the inflammatory response and an increase in immune cell accumulation at the attachment site.19 Immunohistochemical analysis further confirmed the inflammatory microenvironment at the feeding site, showing an influx of inflammatory cells, especially neutrophils, within one hour of TBEV-infected tick feeding. Among these, TBEV antigens were localized in fibroblasts and mononuclear cells, but not in neutrophils.19 These findings suggest that TBEV-infected ticks induce rapid inflammation at the cutaneous interface, potentially affecting the transmission of flaviviruses to hosts. This study contributes to our understanding of the early immunological events during tick-borne flavivirus transmission, emphasizing the significance of localized skin infection in this process (Figure 1).
Figure 1: Proposed overview of the early transmission events of TBEV
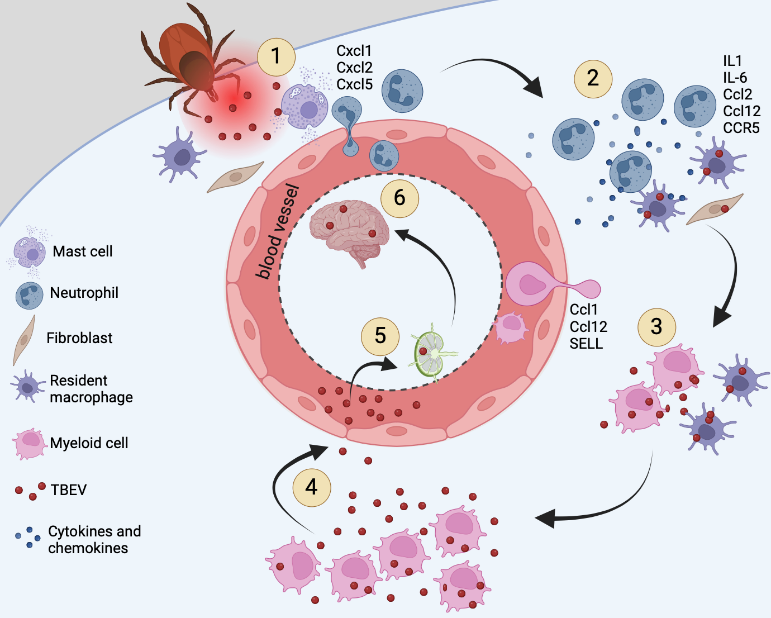
Together these studies illustrate the important role of localized skin infection during the early stages of tick-borne flavivirus transmission.
Neuroinvasion and neurotropism:
Crossing the brain barriers
It is generally believed that neurotropic flaviviruses can invade the CNS by two main routes; the peripheral nervous system or the hematogenous route via the blood. However, the molecular mechanisms governing the neuroinvasion of TBEV and related tick-borne flaviviruses are not yet clear.
Figure 2: Overview of possible routes of TBEV neuroinvasion
The infographic was generated using Biorender (www.biorender.com)
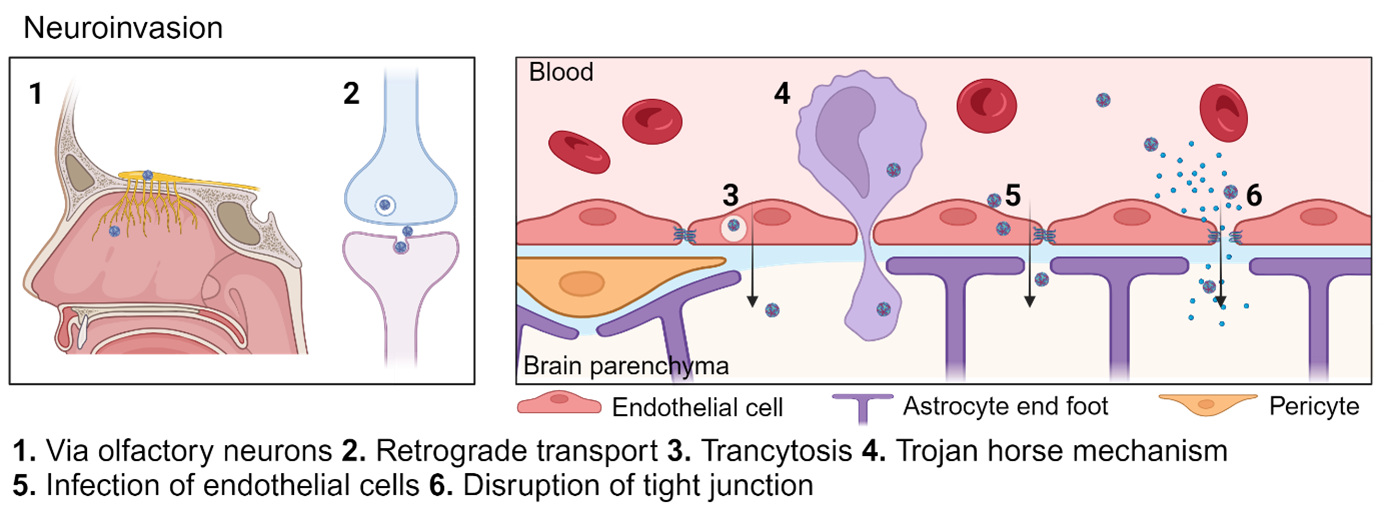
Entry via the peripheral nerves
Some viruses uses the spinal cord to enter the CNS,20,21 however, during experimental infection of TBEV (strain Torö) and LGTV in mice the spinal cord and brain stem are the last infected areas after sub cutaneous (SC) and intraperitoneal (IP) infection respectively.22,23 On the other hand, POWV (LB strain) showed spinal cord infection as early as 4 days post-infection and thereafter a caudal to rostral spread within the brain after high viral dose.24 Indicating that neuroinvasion might depend on the specific virus strain used and the experimental setup. Another report with TBEV (Sofjin) infected mice showed that the autonomic nerves running from the myoenteric plexus were infected as well as the intestine and intestinal lymph nodes after intravenous infection (IV).25 There is direct signaling between the gut to the brain via enteroendocrine cells of the mouse gut that form synapses with vagal neurons26 that may facilitate virus entry. The involvement of the gastrointestinal tract as an important site of infection is supported by the many cases of alimentary TBEV.27-30 However, in mice the oral route of infection is rather ineffective even in highly immunocompromised interferon alpha receptor (IFNAR) knock out mice.31 Infection using oral gavage (with feeding needle) is even less efficient.31 This indicate that the acid environment of the stomach is preventing viral infection, and that the TBEV maybe more likely to establish infection in the mouth or throat. Another possible mechanism for neuroinvasion is via the olfactory sensory neurons in the olfactory bulb. We have seen that the olfactory bulb is the first site of infection after both TBEV (Torö) and LGTV (TP21) after IP and SC infection.22,32 Also supporting this hypothesis is the reported laboratory-acquired infection with TBEV after high titer exposure of aerosols.33 However, since a bi-phasic disease course was observed in this case report it indicates viremia before neuroinvasion,33 and other studies in mice have shown that intranasal infection of mice are less efficient route of infection compared to IP and SC,31,34 thus neuroinvasion via the olfactory neuron seems less likely for TBEV and LGTV.
Hematogenous route of neuroinvasion
The second plausible route of neuroinvasion is the hematogenous via the blood brain barrier (BBB). The BBB is a very tight barrier that separates the blood from the brain parenchyma and the main function is to prevent free diffusion and toxic molecules to enter the brain. The BBB is lining all capillaries in the brain and to prevent permeability and leakage the endothelial cells have tight junctions. These include the claudines and occludin, which are joined to the cytoskeleton by cytoplasmic proteins, such as zonula occludens (ZO).35 Lining the endothelial cells are the pericytes and end-feet from nearby astrocytes, and the crosstalk between endothelia, pericytes and astrocytes are important to preserve the integrity and function of the barrier. For long it was believed that the breakdown of the BBB was important part of neuroinvasion for TBEV as TBE patients show disruption of the BBB.36-38 However, virus is detected the brains of mice days before disruption of the BBB,34,39 and BBB leakage is likely caused by the inflammatory response elicited by the virus in the brain. Microvascular endothelial cells are often used in vitro to mimic the BBB, and infection of these with TBEV (Hypr, Neudoerfl) does not increase permeability or change the key tight junction proteins. Instead the cells become persistently infected and secrete high titers of virus in both directions,40 indicating that TBEV can cross the BBB via a transcellular pathway without changing permeability. In a more complex in vitro model consisting of both human brain endothelial cells and pericytes POWV (LI9, LI41 linage 2 and LB linage 1) infects both cell types persistently and secrets POWV to the lower chamber without changing the permeabilization.41 However, no in vivo experiments have verified infection in the vascular endothelial cells of the BBB. Using single nuclei RNA sequencing Chotiwan et al. recently showed that in the cortex of wt mice the pericytes were infected with LGTV but not endothelial cells.42 The reason for this discrepancy might be that different viral strains and mammalian models were used. Transcytosis is when virus is transported through the cell without productively infecting them. Evidence of transcytosis in vivo through endothelial cells and pericytes has only been shown for Japanese encephalitis (JEV) by electron microscopy.43 Virus could also traffic through the BBB via so called “Trojan horse” mechanism, where virus infected immune cells infiltrate into the brain. However, even though virus infect different immune cells in the periphery, more research is needed to understand the trafficking behavior of infected cells.44
Alternatively, the virus may enter the brain via the blood CSF barrier through the choroid plexus (ChP). ChP is located in the ventricles of the brain and is composed of a monolayer of epithelial cells that contain tight junctions. This epithelial layer rests in a basal lamina surrounding and enclosing a central stroma where dendritic cells, fibroblasts and macrophages can be found. The blood endothelial cells within the ChP central stroma is leaky, thus, the cellular movement of molecules and cells within the CP stroma is not restricted. Both, Zika virus and LGTV have been shown to infect the ChP in vivo, ZIKV targets the pericytes and LGTV targets the ciliated epithelial cells.34,42,45 However, these observations were made in IFNAR knock out mice and not in WT immunocompetent mice, making these observations difficult to translate into TBEV and human situation. Other factors contributing to neuroinvasion in POWV are, the presence of tick saliva,24 active replication in macrophages and prolonged viremia, as resistant mice although with similar peak viremia as susceptible mice clear POWV in the periphery.46
TBEV tropism in the brain
Viral tropism in the brain is determined by several different factors. First the cellular entry receptor is important for binding and viral entry into cells. For TBEV47 and LGTV48 only one entry receptor has been identified, T-Cell Immunoglobulin and Mucin Domain 1 (TIM-1), however it is not likely to be the only one as mice and cells were still susceptible in its absence.47 We have also seen that cellular tropism of infected wt and IFNAR deficient mice with LGTV is markedly different independent of base line expression of the different brain cells,42 indicating that host factors, innate immune response and cellular crosstalk are very important for shaping the cellular tropism in the brain.
After neuroinvasion TBEV targets mainly large neurons of the anterior horns, medulla oblongata, pons, dentate nucleus, Purkinje cells, and striatum in humans.49 Neurons in thalamus, cortex, and Purkinje cells in cerebellum are the main target for TBEV (Hypr) in mice.50 In POWV lineage-1 the main infected areas are brain stem and spinal cord, and the involvement of spinal cord ventral horn and the brain stem might be the cause of the flaccid paralysis in the mice. Infection can also be detected in the cortex, hippocampus and Purkinje cells in cerebellum.51 In LGTV infected rats the virus also infects the Purkinje cells, in addition to infection of midbrain, hippocampus, thalamus and frontal lobe.52 LGTV infection in mice on the other hand does not target the Purkinje cells in the cerebellum but rather excitatory neurons in the entorhinal cortex of the cerebrum.42 Showing that the experimental systems used are very important. The type I IFN response seem to have a major impact on the cellular tropism in vivo. For LGTV, Lindman et al. showed that RIPK3 is important specifically to restrict infection of the granular cell neurons in the cerebellum. This because it is necessary for upregulation of IFNAR expression and thus upregulation of antiviral Interferon stimulated genes (ISGs).53 We have shown that both the specific cells and the areas infected with LGTV in the brain is dependent of type I IFN response.42 In wt mice the excitatory neurons in gray matter of the cerebrum specifically in the entorhinal cortex and audio cortex were infected. Whereas in the absence of IFNAR the tropism shifted to ciliated epithelial cell of the choroid plexus in the ventricles, meninges, and microglia in the white matter tracts of the olfactory.42 The reasons for this dramatic shift in cellular tropism between the mice are likely to be that the cross talk between cells in the brain, and infiltration of immune cells (CD8 T cells expressing IFNγ) into the brain that activates microglia in WT mice by upregulating CCR1. In the absence of IFNAR the crosstalk between cells are blunted, immune cells are not recruited to the brain, and microglia, which expresses high levels of TIM-1 (Human Protein Atlas), are unable to become activated and thus are susceptible to infection.42
Several in vitro studies have shown that primary astrocytes from rat and mouse can be infected with TBEV and they survive and produce virus over many days,54,55 however, in mice TBEV (Hypr) and LGTV is rarely detected in astrocytes.42,50 We have also seen that primary mouse astrocytes cultured in vitro become very susceptible to TBEV (Hypr, Aina and Sofjin) in the absence of IFNAR signaling,56 however, astrocytes are not susceptible in IFNAR knock out mice in vivo,42 indicating that viral tropism studies should be conducted in vivo not in vitro, as cellular tropism of TBF depends on much more than only the entry receptor.
Immune response to TBEV:
Type I interferon response
The type I IFN system is the first line of defense against viral infection and an important part of the intrinsic innate immune response that controls virus dissemination and protects against serious disease. This response rapidly detects invading pathogens and upregulates inhibitory effector proteins and cytokines to ensure survival. The detection of pathogens is based on recognition of the non-self pathogen-associated molecular pattern (PAMP) by specific host sensors, the pattern recognition receptors (PRR). This leads to a signaling cascade and the upregulation and secretion of IFN.57 IFN is a large family of cytokines where the IFNα and -β are type I IFNs and IFNγ is type II IFNs and these are the most studied. Type I IFNs binds to the IFNα receptor (IFNAR), which is expressed on nearly all cell types, in a paracrine and autocrine manner. The IFNAR is composed of a heterodimer of IFNAR1 and IFNAR2. After binding of IFN, the IFNAR activates the Janus kinases, Jak1 and Tyk2, which then phosphorylate the signal transducer and activator of transcription (STAT)-1 and STAT2 proteins, resulting in activation and translocation of the IFN-stimulated gene 3 (ISGF3) transcription factor complex into the nucleus. This ISGF3 induces hundreds of IFN stimulated genes (ISGs), that encode proteins with diverse biological function and some are potent antiviral proteins and part of the response against mammalian viruses.57
Recognition of TBEV and induction of IFN
Rapid detection of the pathogen is crucial for mounting a protective response, and several different PRR families have been identified that recognize numerous ligands. The Toll-like receptors (TLRs) are located on the endosome or the plasma membrane, and the retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs) are in the cytosol. RNA viruses are most likely recognized by TLR3, TLR7, TLR8, or the RLRs (RIG-I and melanoma differentiation-associated gene 5, MDA5), which senses single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA).58-60
For TBEV, it is not totally clear which PRRs are dominant. RIG-I, which recognizes short dsRNA and 5’ PPP, has been shown to be important for IFNβ induction in the U2OS (human osteosarcoma) cell line by siRNA depletion,61 and as MDA5 has been shown to be antagonized by prM of TBEV (Far Eastern subtype) preventing its recruitment to MAVS thus inhibiting IFN upregulation,62 indicating that both are important for sensing. Both RIG-I and MDA5 bind to the adaptor mitochondria-associated IFNβ promoter stimulator-1 (IPS-1, also called MAVS, VISA or CARDIF) via its caspase recruitment domain after binding to its RNA ligand.63 IPS-1 is important for IFNβ induction after TBEV (Hypr) infection in mouse embryonic fibroblasts (MEFs); in its absence, no IFNβ was detected.64 In addition, mice deficient in IPS-1 succumb to LGTV and TBEV (Hypr) infection earlier. These mice showed lower systemic levels of IFNα, resulting in higher viral titers in the periphery and leading to rapid invasion in the CNS.23 IPS-1 is also important in the local IFN response within the brain, reducing viral load and spread of LGTV,23,65,66 indicating an especially important role for RLR in the type I IFN response.
Upon IPS-1 activation, TNF Receptor Associated Factor 3 (TRAF3), TANK Binding Kinase 1 (TBK1) and Inhibitor-κB kinase ε (IKKε) are recruited, leading to phosphorylation and activation of the transcription factor IFN regulatory factor 3 (IRF3). Phosphorylated IRF3, dimerizes and translocate into the nucleus where it binds to the IFNβ gene promoter to initiate transcription and translation.67,68 IFNβ induction after TBEV infection has been shown to be highly dependent on IRF3 activation in the cells, and IRF3 has been shown to dimerize and translocate into the nucleus after TBEV infection.64 However, in vivo type I IFN upregulation is not dependent on IRF3 but on IRF7 in the periphery, and IRF7 plays an important role in the CNS to control infection.69
Very little is known about the importance of TLRs, TLRs signals through the adaptors MyD88 and TRIF, in TBEV infection. In vitro, most cells do not upregulate IFNβ in the absence of IPS1 in vitro.64 A recent study, however, showed that astrocytes depend on TLR sensing and MyD88/TRIF for the upregulation of IFNs later during infection and that TLRs are upregulated by TBEV (Neudoerfl).70 It also seems that the TLR7 is more important for regulating neuroinflammation than type I IFNs.71 As mice deficient in TLR7 have higher viral load in the CNS and lower levels of pro-inflammatory cytokines. Primary neurons did not show a difference in infection rate, but TLR7 deficient neurons induced higher levels of IFNβ.71 A functional TLR3 has been associated with TBE in humans.72
Figure 3: Viral evasion of IFN induction
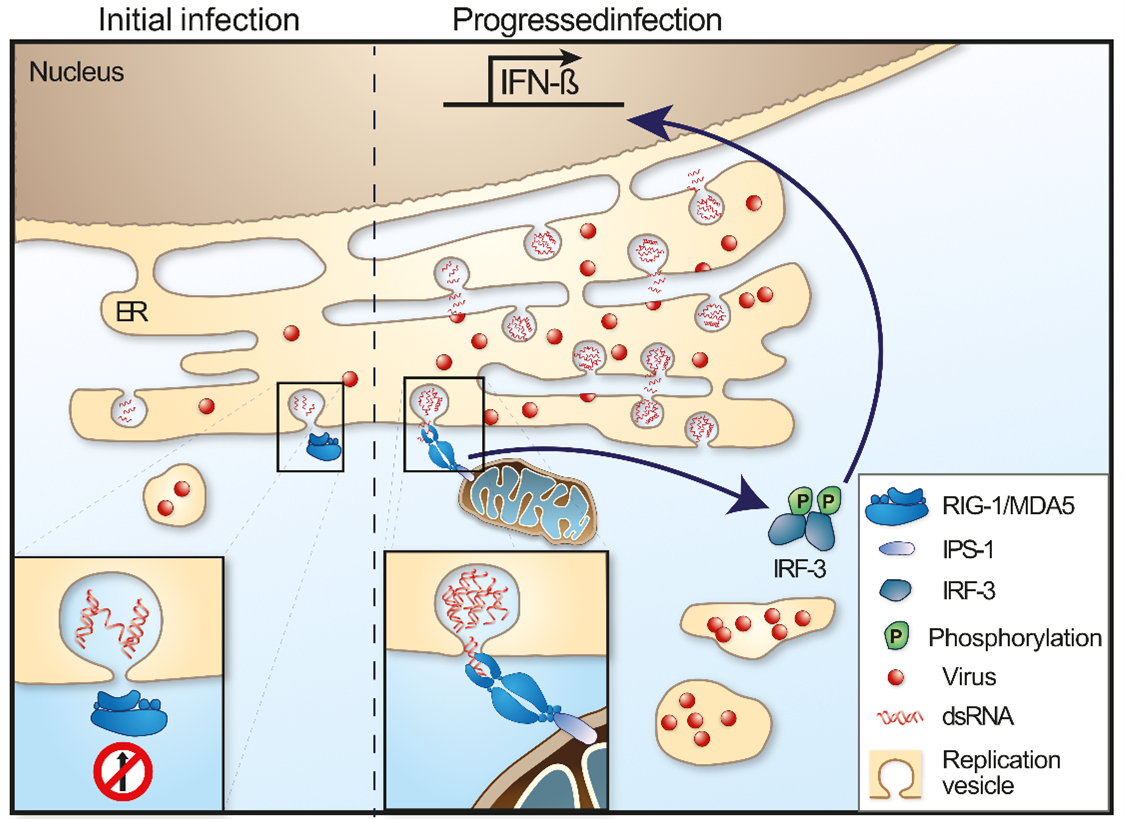
Since the type I IFN response is so important in controlling and restricting viral replication, most viruses have developed strategies to prevent upregulation of IFN by antagonizing the different steps in the IFN induction pathway.74-76 For TBEV (Far Eastern subtype) the prM was recently identified to prevent interaction and signaling between MDA5 and MAVS.62 TBEV also employ a passive escape mechanism that delays the induction of IFNβ by replicating inside replication vesicles or packets, thereby hiding its dsRNA from RIG-I and other PRRs (Figure 3).61,64,73,77 Later, during infection, the dsRNA leaks out from the replication vesicles, IRF3 is activated and translocates into the nucleus to transcribe IFNβ, which then is translated and secreted. Thus, the virus is produced and released from the cell before IFNβ can trigger an antiviral response in neighboring cells (Figure 3).64,73 Interestingly, different cell types respond to infection in different ways with different kinetic. Primary mouse astrocytes have a very fast type I IFN response and secret IFNs that can protect, astrocytes and primary cortical neurons in culture already 3 to 6 h post infection,56 and also co-cultured neurons.78
Type I IFN signaling and response against TBEV
After infection and secretion of IFN, the IFN binds to its receptor the IFNAR1/2 which stimulates the upregulation of hundreds of ISGs that can limit the infection. The ISGs encode for PRR, adaptors and transcription factors to ensure a rapid response after infection. Cytokines and chemokines are also produced which activate and recruit immune cells to limit the infection, as well as antiviral proteins that can target viral replication directly in the cell.79 The IFNAR is therefore a key molecule in the type I IFN response. The importance of this molecule has been demonstrated for many viruses. For LGTV the type I IFN response determines tropism and can protect mice from lethal infection. In the absence of this response, the virus replicates uncontrollably in all organs, induces a rapid opening of the blood-brain barrier, and the mice succumb very quickly. This research also has shown that IFNAR is important in all cell types; hematopoietic, stroma, neuroectodermal and cells in the periphery.34
Most steps in the viral “life” cycle are targeted by 1 or several antiviral proteins encoded by the ISGs. Several ISGs have been identified to have antiviral effect on TBEV the Interferon-induced transmembrane proteins (IFITMs) 1, 2, 3, the rodent tripartite motif (TRIM) protein, TRIM79α, and viperin (virus inhibitory protein, endoplasmic reticulum-associated, IFN-inducible).80-82 Although all three IFITM proteins are antivirally active IFITM3 is the most potent one and can protect against virus induced cell death, and IFITM proteins are most effective against cell free virus and not against cell to cell virus spread.80 The antiviral mechanism of TRIM79α is direct targeting of the viral polymerase, the non-structural protein 5 (NS5), an essential component of the replication complex, for lysosomal degradation. TRIM79α seems to be specific for TBEV and LGTV, because mosquito-borne flaiviviruses; WNV and Japanese encephalitis virus (JEV), were shown not to be restricted by this protein.81
Viperin, on the other hand, is a highly conserved protein with broad spectrum antiviral activity, which has been shown to restrict a diverse range of viruses from different families. For the Flaviviridae family, viperin restricts hepatitis C, DENV, WNV and TBEV. However, the antiviral mechanism seem to depend on the specific virus. For TBEV, viperin selectively target the positive stranded RNA synthesis. The intracellular location to the ER via viperins N-terminal amphipathic alpha helix is important as it coincides with viral replication. The antiviral activity is depending on the radical S-adenosyl methionine (SAM) domain and the proper iron-sulphur maturation of the protein.82,83 Recent studies have identified several viral and cellular interaction partners to viperin.32,83-87 Viperin is able to target TBEV in multiple ways mediating antiviral activity in a cell type-specific manner. Viperin interacts with several TBEV proteins; prM, E, NS2A, NS2B and NS3. The interaction between NS3 and viperin results in proteasome-dependent degradation of NS3.86 The stability of prM, E, NS2A and NS2B are affected by viperin, but only in the presence of NS3.86 Interestingly, although viperin do not directly interact with the TBEV C protein, viperin expression induce C particle formation and release from virus infected cells and disturbing the assembly process of TBEV.87 Viperin mediates this effect by interacting and sequestering the cellular protein Golgi brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1),87 which is involved in the vesicular trafficking of the secretory pathway88,89 and is a pro-viral factor for many different viruses.90-93 Thus, viperin may target other viruses via its interaction with GBF1. The in vivo importance of viperin during TBEV infection was recently shown in the viperin-/- mice.32 This study show that specific regions of the brain rely differentially on the antiviral activity of viperin for protection against LGTV. Viperin is important in the olfactory bulb and cerebrum, while viral replication were unchanged in cerebellum and brain stem in the absence of viperin. This effect is due to the different neuronal subtypes, viperin expression is very important in cortical neurons but not at all in granular cell neurons isolated from the cerebellum.32 Looking at polymorphisms in human TBE have identified several ISGs associated with TBE disease for example Interferon Induced Protein With Tetratricopeptide Repeats 1 (IFIT1),94 2′-5′-oligoadenylate synthetase (OAS)2 and OAS3.95,96
Figure 4: Interferon signaling and inhibition
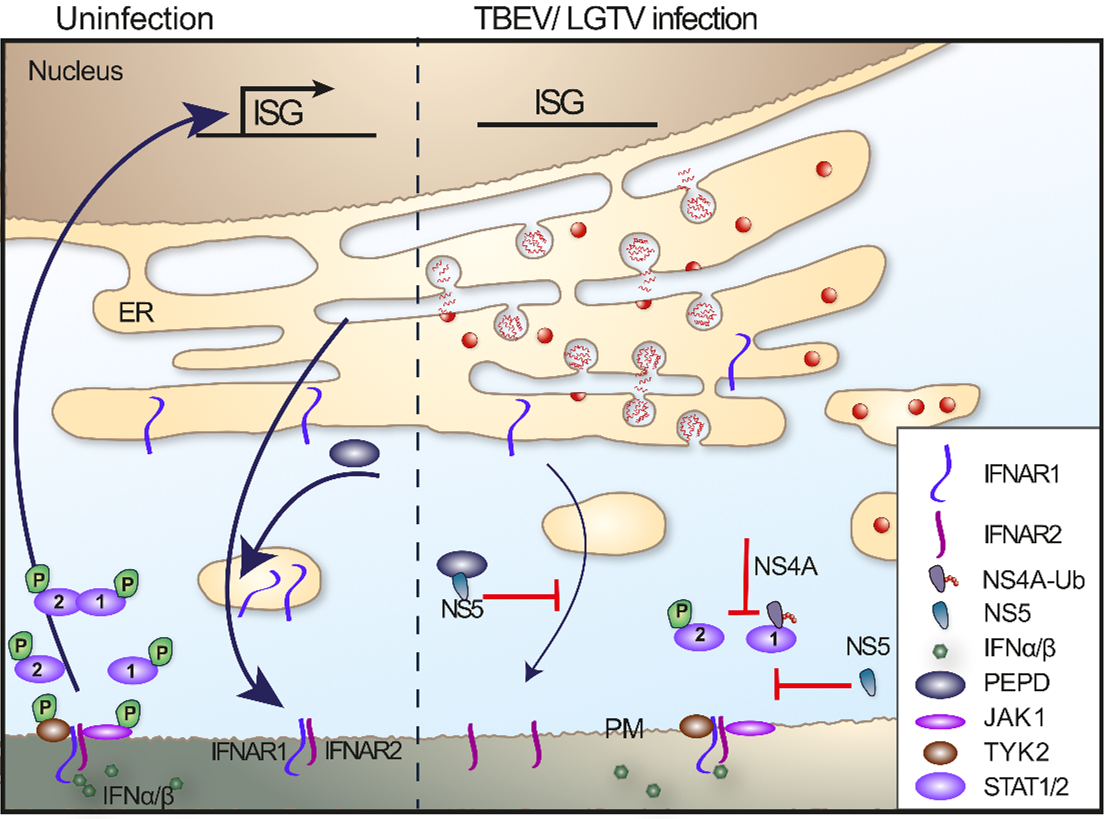
Even though different ISGs can potently restrict TBEV replication if induced before infection,56,81,82,98 IFN treatment after infection has limited effect in vitro.98 The reason for this is the expression of an IFN antagonist, NS4A100 and NS5.98,99 TBEV NS4A blocks the phosphorylation and dimerization of STAT1/STAT2 to reduce the type I and type II IFN-mediated signaling.100 The NS5 protein of LGTV interferes with the phosphorylation of Jak1 and Tyk2 in response to IFNβ, which leads to failure of STAT1/2 phosphorylation and subsequent ISG expression.98,99 Werme et al. showed that the interaction between Scribble and NS5 is important for plasma membrane targeting and IFN antagonist activity; however, the exact target of NS5 is unclear.99 In addition, NS5 was shown to block IFN signaling by selectively reducing the level of IFNAR1 expression on the cell surface. This reduction was dependent on NS5 binding to prolidase. Prolidase is needed for IFNAR1 intracellular trafficking, maturation, activation of IFNβ-stimulated gene induction, and IFN-I-dependent viral control (Figure 4).97 The relationship between NS5 function and virulence has not been observed for tick-borne flaviviruses, such as TBEV and the low virulence LGTV NS5; both exhibited the same degree of p-STAT inhibition. However, there are most likely other viral proteins that are important for pathogenicity and suppression of innate immune responses, as this has been shown for other flaviviruses. However, for TBEV these mechanisms have yet to be identified.
Adaptive immune response against TBEV
Humoral immunity is an important component of the immune response. As with other flaviviruses, a functional humoral immune response is critically important in controlling infections.101 Depleting B cells with immunosuppressive treatment of Rituximab lead to severe and fatal TBE.102 On the other hand, passive transfer of monoclonal or polyclonal TBEV-specific antibodies protects mice in vivo and protection correlates with in vitro neutralization.103-107 No infectious virus could be detected in the blood or brain of passively protected mice subsequent to TBEV challenge. However, in a vaccination study the antibodies response protected against disease but did not from neuroinvasion, as viral RNA was detected in the CNS.50 However, antibodies protect not only by neutralization; therefore, because limited virus replication does occur, this indicates that mechanisms of protection from disease exist other than sterilizing immunity.108
In addition to effective humoral immunity, the activation of cellular immunity is usually required for clearance of established infection. Distinct T cell subsets play a key role in the induction of protective immune response against TBEV infections. CD4+ T cells are essential in priming the TBEV-specific antibody response and sustaining the CD8+ T cell response.
For more details about the interplay between TBEV and the humoral immune response, cellular immune response, and different innate immune cells please visit Chapter 7 Immunology of TBEV infection by Zens and Ackermann-Gäumann.
Tools to study pathogenesis:
Overview of relevant animal models
Animal models are pivotal in comprehending the pathogenesis, transmission dynamics, and potential interventions for tick-borne encephalitis virus infection. An optimal animal model should closely emulate the human condition in terms of disease symptoms and underlying mechanisms. Tick-borne viruses exhibit minimal host specificity due to ticks’ feeding habits, which vary as they mature and can encompass hosts of various sizes or species without preference. Humans typically become infected incidentally when ticks venture beyond their natural habitats or human ventures into the habitat of ticks. The diverse array of hosts that ticks can feed on renders many tick-borne viruses amenable to investigation using laboratory animals.
Both large and small animal models have been utilized to explore the fundamental aspects of TBEV infection, disease progression, and neuropathogenesis. Early investigations in sheep resulted in a better understanding of the differential neurovirulence and pathogenesis of TBEV.109 Several species of non-human primates, such as Macaca mulatta (rhesus macaques), Cercopithecus aethiops (African Green monkeys), Macaca fascicularis (Crab-eating macaques), Macaca cynomolgus, and Macaca sylvanus, have been employed to study TBE neuropathogenesis. Though non-human primate models do not mimic human clinical outcomes, they are a good model to understand TBEV infections and to evaluate vaccine efficacy.110-113
Small mammals such as Syrian golden hamsters,114 moles115 have been used to understand TBEV pathogenesis and disease progression. However, they show reduced susceptibility. Laboratory mice such as ICR, C57BL/6 or BALB/c mice serve as a promising animal model for advancing research into the mechanisms underlying tick-borne virus infections and their pathogenesis.22,116-120 Due to their closer phylogenetic relationship with humans and notable genomic similarities, especially evident in knock-out mice, where specific genes are deleted to elucidate mammalian genetic factors in infection and disease progression, they offer valuable insights.23,119 Mice are susceptible to TBEV isolates, resulting in fever and neurological symptoms resembling human encephalitis. Histological examination of infected mice has unveiled substantial brain inflammation and damage, aligning with clinical manifestations observed in human cases.116,117,119,120
Kurhade et al. (2018) used C57BL/6 mice to characterize the pathogenesis of TBEV isolated from 2 different transmission foci.22 The investigators compared the neuroinvasiveness, neurovirulence, and immune response of two European strains (HB171/11 from Germany and Toro-2003 from Sweden) in mice, uncovering distinct differences that enhance our understanding of TBEV pathogenesis. The HB171/11 is low virulent tick isolate from a focus where TBE patients only show gastrointestinal and constitutional symptoms.121 The Torö-2003 strain is an infectious clone from an island where 32 neurological TBE cases122 occurred. The strain HB171/11 was found to be a low virulent phenotype with low or delayed neuroinvasiveness, and the Toro-2003 strain was found to be highly pathogenic.22
In addition, mice have also been used to investigate viral genetic determinants of infection and pathogenesis, and E protein, NS2B, NS3, NS5 protein, and the variable region of the 3’ untranslated region have been shown to be important for determining pathogenicity in mice.118,122-127 However, more studies are needed to fully understand the reason for the different clinical outcomes. Some strains of TBEV and POWV have been suggested to become persistent or chronic however, the mechanism is not clear, but it is interesting that in experimental models of TBEV and related viruses, the virus RNA is found in the brain of rodents128-132 and in non-human primates110,113,133,134 for a long time even in the absence of severe disease in the acute phase, although it is not clear if the virus RNA is infectious.
The variety of animal models utilized in research on TBEV underscores the comprehensive strategy needed to grasp and fight this virus, with mice being pivotal in revealing the mechanisms of infection and the progression of the disease.
Reverse genetics systems
Reverse genetics of viruses is the generation and manipulation of viral genomes to investigate the direct effects of changes on virus biology and pathogenesis. For flaviviruses, the first reverse genetic system was developed in 1989 for YFV.135 Since the genome of flaviviruses is positive stranded, they are infectious if introduced into susceptible cells.136 There are several different approaches to generate infectious virus. One important step is the generation of a complementary DNA (cDNA) to the RNA genome. The cDNA is often cloned into a plasmid under a specific promoter, which enables the in vitro transcription of viral RNA. This DNA clone enables the introduction of mutations into the genome, and subsequent analysis of the resulting phenotype. Reverse genetics have been used to study virulence, replication, host range, vaccines, and functions of the coding and non-coding regions. However, these clones are laborious and difficult to generate due to instability and toxicity of some viral sequences in bacteria.137
For TBEV 2 separate approaches were used in the beginning; plasmid-based infectious clones138 and the PCR based methods for constructing recombinant virus.139,140 Both rely on in vitro transcription and transfection of RNA. The most recent technique for generating TBEV clones is the infectious-subgenomic-amplicon (ISA) method. Three PCR amplicons are produced that have a CMV promoter at the 5′ non-coding region (NCR) and 70-100 bp overlapping regions; the hepatitis delta ribozyme is followed by the simian virus 40 polyadenylation signal. The amplicons are mixed and introduced into the cells where they recombine and produce infectious virus.141
Infectious clone systems have been very useful in studying determinants of replication and biological characteristics as well as to identify pathogenicity factors of TBEV. Two advantages of this approach are that the genome is defined and can be manipulated. In contrast, natural viral isolates of positive stranded RNA viruses are present as a population of different viral types also called quasispecies. This is due to the error prone RNA dependent RNA polymerase. In addition, manipulating natural viral isolates with specific mutagenesis inducing drugs is a very nonspecific approach.
With this technique, several determinates of pathogenicity have been identified. Specifically, the envelope protein responsible for receptor mediated entry,126 the function of the membrane protein in virus budding,142 and the importance of different regions in the 3’NCR. Neurovirulence in mice was shown to be dependent on specific amino acid residues in the upper lateral surface of domain III in the envelope (E) protein of TBEV (residues E308, E310 and E311), possibly due to disruption of the receptor binding.126 The residues S267L, K315E, N389D in LGTV E protein and K46E in the NS3 protein, were shown to be crucial for neuroinvasiveness in immunodeficient mice.143 The 5’ and the 3’ NCR contain complementary sequences that help genomic cyclization to form panhandle structures. The NCRs have several conserved structural stem loops that are important for replication, translation initiation and packaging.144,145 At the beginning of the flavivirus 3’ NCR, a secondary structure forms a pseudoknot that protects the terminal 300 to 500 bases from exoribonuclease XRN1 degradation, generating a subgenomic flavivirus RNA (sfRNA).146-148 The sfRNA has been shown to be critical for WNV induced cytopathic effects149 and pathogenicity in mice,149 and is involved in viral subversion of type I IFN response by a yet unknown mechanism.150 The TBEV sfRNA has been shown to specifically interfere with the RNAi system of ticks.151 The 3’ NCR of TBEV can be divided into a highly conserved core element and a variable region that is both heterogenic in length and sequence.152 Several European TBEV strains contain an internal poly(A) tract in the variable region of the 3’ NCR, which was considered dispensable for replication and virulence in mice.127,153 However, studies recently showed that the variable region and the poly(A) tract can modulate virulence of the Far Eastern TBEV.123,154 We have also detected different lengths of the poly(A) tract in a blood feeding tick indicating that the poly(A) might be important for the switch between invertebrate to vertebrate.155 To investigate this further a long poly(A) Torö-38A and a TBEV Torö with a short poly(A) were cloned and rescued. We were able to show that the viruses with long poly(A) were attenuated in cell culture but more virulent in mice compared with the short poly(A), and the genome with short poly(A) was much more stable compared with the long version, which developed a high quasispecies diversity.122
Ongoing challenges and areas for future investigation
Important advances in the identification of molecular and cellular mechanisms of TBEV-induced pathogenesis have been made in recent years. Skin is the interface between a feeding TBEV-infected tick and a host; consequently, the cutaneous immune cells likely play a crucial role in virus transmission. In the earliest stages of TBEV-infected tick feeding, a complex, inflammatory micro-environment exists in the mammalian host’s skin, with increased recruitment, migration, and accumulation of Langerhans cells, mononuclear phagocytes, and neutrophils. The dynamic secretion of tick salivary factors at the infected tick feeding foci modulates the cutaneous micro-environment to facilitate TBEV transmission, establishment, and dissemination from the skin to the terminal organs. However, many unanswered questions remain about the function of immune cells at the feeding site of a TBFV-infected tick. Modern single-cell and spatial transcriptomics techniques will allow us to investigate these early transmission events. They will enable us to understand immune processes at a single-cell level. In addition, gaps exist in our current understanding of the dissemination of viruses from the skin to the central nervous system. A better understanding of the virus transmission, establishment, neuroinvasion, dissemination and cellular tropism within the brain will allow us to develop novel countermeasures to prevent TBEV transmission, treat TBEV infections, and reduce disease burden. The interactions between the virus and the innate and adaptive immune response are not fully understood. The use of reverse genetics, specific knock out mouse models, new technologies like whole brain imaging, single cell sequencing and spatial transcriptomics will greatly advance our understanding of TBEV pathogenesis in the future.
Contact
Anna K Överby
Anna.overby@umu.se
Affiliations
Anna K Överby, Saravanan Thangamani
Citation
Överby AK, Thangamani S. Pathogenesis of TBEV-diseases. Chapter 6. In: Dobler G, Erber W, Bröker M, Chitimia-Dobler L, Schmitt HJ, eds. The TBE Book. 7th ed. Singapore: Global Health Press; 2024. doi:10.33442/26613980_6-7
References
- Gritsun TS, Nuttall PA, Gould EA. Tick-borne flaviviruses. Advances in virus research. 2003;61:317-71.
- Gritsun TS, Lashkevich VA, Gould EA. Tick-borne encephalitis. Antiviral Res. Jan 2003; 57(1-2):129-46.
- Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet. May 31 2008;371(9627):1861-71.
- Kovalev SY, Mukhacheva TA. Tick-borne encephalitis virus subtypes emerged through rapid vector switches rather than gradual evolution. Ecol Evol. Nov 2014;4(22):4307-16. doi:10.1002/ece3.1301
- Süss J. Tick-borne encephalitis 2010: epidemiology, risk areas, and virus strains in Europe and Asia-an overview. Ticks Tick Borne Dis. Mar 2011;2(1):2-15. doi:10.1016/j.ttbdis.2010.10.007
- Danielová V, Holubová J, Pejcoch M, Daniel M. Potential significance of transovarial transmission in the circulation of tick-borne encephalitis virus. Folia Parasitol (Praha). 2002;49(4):323-5.
- Alekseev AN, Burenkova LA, Vasilieva IS, Dubinina HV, Chunikhin SP. Preliminary studies on virus and spirochete accumulation in the cement plug of ixodid ticks. Exp Appl Acarol. Dec 1996;20(12):713-23. doi:10.1007/bf00051556
- Ebel GD, Kramer LD. Short report: duration of tick attachment required for transmission of powassan virus by deer ticks. Am J Trop Med Hyg. Sep 2004;71(3):268-71.
- Nuttall PA, Labuda M. Tick-host interactions: saliva-activated transmission. Parasitology. 2004;129 Suppl:S177-89. doi:10.1017/s0031182004005633
- Labuda M, Jones LD, Williams T, Nuttall PA. Enhancement of tick-borne encephalitis virus transmission by tick salivary gland extracts. Med Vet Entomol. Apr 1993;7(2):193-6.
- Labuda M, Jones LD, Williams T, Danielova V, Nuttall PA. Efficient transmission of tick-borne encephalitis virus between cofeeding ticks. J Med Entomol. Jan 1993;30(1):295-9. doi:10.1093/jmedent/30.1.295
- Randolph SE. Transmission of tick-borne pathogens between co-feeding ticks: Milan Labuda’s enduring paradigm. Ticks Tick Borne Dis. Dec 2011;2(4):179-82. doi:10.1016/j.ttbdis.2011.07.004
- Kazimirova M, Thangamani S, Bartikova P, et al. Tick-Borne Viruses and Biological Processes at the Tick-Host-Virus Interface. Front Cell Infect Microbiol. 2017;7:339. doi:10.3389/fcimb.2017.00339
- Labuda M, Austyn JM, Zuffova E, et al. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology. May 15 1996;219(2):357-66. doi:10.1006/viro.1996.0261
- Lieskovská J, Páleníková J, Langhansová H, Chmelař J, Kopecký J. Saliva of Ixodes ricinus enhances TBE virus replication in dendritic cells by modulation of pro-survival Akt pathway. Virology. Jan 15 2018;514:98-105. doi:10.1016/j.virol.2017.11.008
- Nuttall PA, Labuda M. Dynamics of infection in tick vectors and at the tick-host interface. Advances in virus research. 2003;60:233-72. doi:10.1016/s0065-3527(03)60007-2
- Johnston LJ, Halliday GM, King NJ. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J Invest Dermatol. Mar 2000;114(3):560-8. doi:10.1046/j.1523-1747.2000.00904.x
- Fialova A, Cimburek Z, Iezzi G, Kopecky J. Ixodes ricinus tick saliva modulates tick-borne encephalitis virus infection of dendritic cells. Microbes and infection / Institut Pasteur. Jul 2010;12(7):580-5. doi:10.1016/j.micinf.2010.03.015
- Thangamani S, Hermance ME, Santos RI, et al. Transcriptional Immunoprofiling at the Tick-Virus-Host Interface during Early Stages of Tick-Borne Encephalitis Virus Transmission. Front Cell Infect Microbiol. 2017;7:494. doi:10.3389/fcimb.2017.00494
- Hixon AM, Clarke P, Tyler KL. Contemporary Circulating Enterovirus D68 Strains Infect and Undergo Retrograde Axonal Transport in Spinal Motor Neurons Independent of Sialic Acid. Journal of virology. Aug 15 2019;93(16). doi:10.1128/jvi.00578-19
- Chen CS, Yao YC, Lin SC, et al. Retrograde axonal transport: a major transmission route of enterovirus 71 in mice. Journal of virology. Sep 2007;81(17):8996-9003. doi:10.1128/jvi.00236-07
- Kurhade C, Schreier S, Lee YP, et al. Correlation of Severity of Human Tick-Borne Encephalitis Virus Disease and Pathogenicity in Mice. Emerg Infect Dis. Sep 2018;24(9):1709-1712. doi:10.3201/eid2409.171825
- Kurhade C, Zegenhagen L, Weber E, et al. Type I Interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by IPS-1. J Neuroinflammation. 2016;13(1):22. doi:10.1186/s12974-016-0487-9
- Santos RI, Hermance ME, Reynolds ES, Thangamani S. Salivary gland extract from the deer tick, Ixodes scapularis, facilitates neuroinvasion by Powassan virus in BALB/c mice. Sci Rep. Oct 22 2021;11(1):20873. doi:10.1038/s41598-021-00021-2
- Nagata N, Iwata-Yoshikawa N, Hayasaka D, et al. The pathogenesis of 3 neurotropic flaviviruses in a mouse model depends on the route of neuroinvasion after viremia. Journal of neuropathology and experimental neurology. Mar 2015;74(3):250-60. doi:10.1097/NEN.0000000000000166
- Kaelberer MM, Buchanan KL, Klein ME, et al. A gut-brain neural circuit for nutrient sensory transduction. Science. Sep 21 2018;361(6408). doi:10.1126/science.aat5236
- Buczek AM, Buczek W, Buczek A, Wysokińska-Miszczuk J. Food-Borne Transmission of Tick-Borne Encephalitis Virus-Spread, Consequences, and Prophylaxis. Int J Environ Res Public Health. Feb 5 2022;19(3). doi:10.3390/ijerph19031812
- Gonzalez G, Bournez L, Moraes RA, et al. A One-Health Approach to Investigating an Outbreak of Alimentary Tick-Borne Encephalitis in a Non-endemic Area in France (Ain, Eastern France): A Longitudinal Serological Study in Livestock, Detection in Ticks, and the First Tick-Borne Encephalitis Virus Isolation and Molecular Characterisation. Front Microbiol. 2022;13:863725. doi:10.3389/fmicb.2022.863725
- Kerlik J, Avdičová M, Štefkovičová M, et al. Slovakia reports highest occurrence of alimentary tick-borne encephalitis in Europe: Analysis of tick-borne encephalitis outbreaks in Slovakia during 2007-2016. Travel Med Infect Dis. Nov-Dec 2018;26:37-42. doi:10.1016/j.tmaid.2018.07.001
- Ličková M, Fumačová Havlíková S, Sláviková M, Klempa B. Alimentary Infections by Tick-Borne Encephalitis Virus. Viruses. Dec 30 2021;14(1). doi:10.3390/v14010056
- Schreier S, Cebulski K, Kröger A. Contact-dependent transmission of Langat and tick-borne encephalitis virus in type I interferon receptor-1 deficient mice. Journal of virology. Mar 25 2021;95(8). doi:10.1128/jvi.02039-20
- Lindqvist R, Kurhade C, Gilthorpe JD, Overby AK. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J Neuroinflammation. Mar 15 2018;15(1):80. doi:10.1186/s12974-018-1119-3
- Avsic-Zupanc T, Poljak M, Maticic M, et al. Laboratory acquired tick-borne meningoencephalitis: characterisation of virus strains. Clin Diagn Virol. Jul 1995;4(1):51-9. doi:10.1016/0928-0197(94)00062-y
- Weber E, Finsterbusch K, Lindquist R, et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. Journal of virology. Nov 2014;88(21):12202-12. doi:10.1128/JVI.01215-14
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. Sep 2006;1(3):223-36. doi:10.1007/s11481-006-9025-3
- Chekhonin VP, Zhirkov YA, Belyaeva IA, Ryabukhin IA, Gurina OI, Dmitriyeva TB. Serum time course of two brain-specific proteins, alpha(1) brain globulin and neuron-specific enolase, in tick-born encephalitis and Lyme disease. Clin Chim Acta. Jun 2002;320(1-2):117-25. doi:10.1016/s0009-8981(02)00057-8
- Kang X, Li Y, Wei J, et al. Elevation of matrix metalloproteinase-9 level in cerebrospinal fluid of tick-borne encephalitis patients is associated with IgG extravassation and disease severity. PLoS One. 2013;8(11):e77427. doi:10.1371/journal.pone.0077427
- Moniuszko A, Pancewicz S, Czupryna P, et al. ssICAM-1, IL-21 and IL-23 in patients with tick borne encephalitis and neuroborreliosis. Cytokine. Nov 2012;60(2):468-72. doi:10.1016/j.cyto.2012.05.007
- Ruzek D, Salat J, Singh SK, Kopecky J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One. 2011;6(5):e20472. doi:10.1371/journal.pone.0020472
- Palus M, Vancova M, Sirmarova J, Elsterova J, Perner J, Ruzek D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology. Jul 2017;507:110-122. doi:10.1016/j.virol.2017.04.012
- Conde JN, Sanchez-Vicente S, Saladino N, et al. Powassan Viruses Spread Cell to Cell during Direct Isolation from Ixodes Ticks and Persistently Infect Human Brain Endothelial Cells and Pericytes. Journal of virology. Jan 12 2022;96(1):e0168221. doi:10.1128/jvi.01682-21
- Chotiwan N, Rosendal E, Willekens SMA, et al. Type I interferon shapes brain distribution and tropism of tick-borne flavivirus. Nat Commun. Apr 10 2023;14(1):2007. doi:10.1038/s41467-023-37698-0
- Liou ML, Hsu CY. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. Sep 1998;293(3):389-94. doi:10.1007/s004410051130
- Marshall EM, Koopmans MPG, Rockx B. A Journey to the Central Nervous System: Routes of Flaviviral Neuroinvasion in Human Disease. Viruses. Sep 21 2022;14(10). doi:10.3390/v14102096
- Kim J, Alejandro B, Hetman M, et al. Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood-cerebrospinal fluid barrier. PLoS pathogens. May 2020;16(5):e1008204. doi:10.1371/journal.ppat.1008204
- Jasperse BA, Mattocks MD, Noll KE, Ferris MT, Heise MT, Lazear HM. Neuroinvasive Flavivirus Pathogenesis Is Restricted by Host Genetic Factors in Collaborative Cross Mice, Independently of Oas1b. Journal of virology. Jul 27 2023;97(7):e0071523. doi:10.1128/jvi.00715-23
- Zhang X, Liang C, Wang H, et al. T-Cell Immunoglobulin and Mucin Domain 1 (TIM-1) Is a Functional Entry Factor for Tick-Borne Encephalitis Virus. mBio. Jan 25 2022;13(1):e0286021. doi:10.1128/mbio.02860-21
- Rodrigues R, Danskog K, Overby AK, Arnberg N. Characterizing the cellular attachment receptor for Langat virus. PLoS One. 2019;14(6):e0217359. doi:10.1371/journal.pone.0217359
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. Journal of neuropathology and experimental neurology. Jun 2005;64(6):506-12.
- Petry M, Palus M, Leitzen E, et al. Immunity to TBEV Related Flaviviruses with Reduced Pathogenicity Protects Mice from Disease but Not from TBEV Entry into the CNS. Vaccines. Feb 26 2021;9(3). doi:10.3390/vaccines9030196
- Santos RI, Hermance ME, Gelman BB, Thangamani S. Spinal Cord Ventral Horns and Lymphoid Organ Involvement in Powassan Virus Infection in a Mouse Model. Viruses. Aug 12 2016;8(8). doi:10.3390/v8080220
- Maffioli C, Grandgirard D, Engler O, Leib SL. A tick-borne encephalitis model in infant rats infected with langat virus. Journal of neuropathology and experimental neurology. Dec 2014;73(12):1107-15. doi:10.1097/nen.0000000000000131
- Lindman M, Angel JP, Estevez I, et al. RIPK3 promotes brain region-specific interferon signaling and restriction of tick-borne flavivirus infection. PLoS pathogens. Nov 2023;19(11):e1011813. doi:10.1371/journal.ppat.1011813
- Palus M, Bily T, Elsterova J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. The Journal of general virology. Nov 2014;95(Pt 11):2411-26. doi:10.1099/vir.0.068411-0
- Potokar M, Jorgačevski J, Zorec R. Astrocytes in Flavivirus Infections. Int J Mol Sci. Feb 6 2019;20(3). doi:10.3390/ijms20030691
- Lindqvist R, Mundt F, Gilthorpe JD, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation. Oct 24 2016;13(1):277. doi:10.1186/s12974-016-0748-7
- Weber F, Kochs G, Haller O. Inverse interference: how viruses fight the interferon system. Viral Immunol. 2004;17(4):498-515.
- Nazmi A, Dutta K, Hazra B, Basu A. Role of pattern recognition receptors in flavivirus infections. Virus research. Jun 24 2014;185:32-40. doi:10.1016/j.virusres.2014.03.013
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunological reviews. Jan 2009;227(1):54-65.
- Akira S, Takeda K. Toll-like receptor signalling. Nature reviews. Jul 2004;4(7):499-511. doi:10.1038/nri1391
- Miorin L, Albornoz A, Baba MM, D’Agaro P, Marcello A. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus research. Feb 2012;163(2):660-6. doi:10.1016/j.virusres.2011.11.020
- Sui L, Zhao Y, Wang W, et al. Flavivirus prM interacts with MDA5 and MAVS to inhibit RLR antiviral signaling. Cell Biosci. Jan 13 2023;13(1):9. doi:10.1186/s13578-023-00957-0
- Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature immunology. Oct 2005;6(10):981-8. doi:ni1243 [pii] 10.1038/ni1243
- Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. Journal of virology. Sep 2010;84(17):8470-83. doi:10.1128/jvi.00176-10
- Zegenhagen L, Kurhade C, Koniszewski N, Overby AK, Kroger A. Brain heterogeneity leads to differential innate immune responses and modulates pathogenesis of viral infections. Cytokine & growth factor reviews. 2016. doi:10.1016/j.cytogfr.2016.03.006
- Zegenhagen L, Kurhade C, Kroger A, Overby AK. Differences in IPS-1 mediated innate immune responses between neurotrophic flavivirus infection. Journal of Neuroinfectious Diseases. 2016;7(210). doi:10.4172/2314-7326.1000210
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. The Journal of biological chemistry. May 25 2007;282(21):15325-9.
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. The EMBO journal. Feb 16 1998;17(4):1087-95.
- Weichert L, Düsedau HP, Fritzsch D, et al. Astrocytes evoke a robust IRF7-independent type I interferon response upon neurotropic viral infection. J Neuroinflammation. Sep 22 2023;20(1):213. doi:10.1186/s12974-023-02892-w
- Ghita L, Breitkopf V, Mulenge F, et al. Sequential MAVS and MyD88/TRIF signaling triggers anti-viral responses of tick-borne encephalitis virus-infected murine astrocytes. J Neurosci Res. Oct 2021;99(10):2478-2492. doi:10.1002/jnr.24923
- Baker DG, Woods TA, Butchi NB, et al. Toll-like receptor 7 suppresses virus replication in neurons but does not affect viral pathogenesis in a mouse model of Langat virus infection. The Journal of general virology. Feb 2013;94(Pt 2):336-47. doi:10.1099/vir.0.043984-0 vir.0.043984-0 [pii]
- Kindberg E, Vene S, Mickiene A, Lundkvist A, Lindquist L, Svensson L. A functional Toll-like receptor 3 gene (TLR3) may be a risk factor for tick-borne encephalitis virus (TBEV) infection. J Infect Dis. 2011;203(4):523-528. doi:10.1093/infdis/jiq082
- Overby AK, Weber F. Hiding from intracellular pattern recognition receptors, a passive strategy of flavivirus immune evasion. Virulence. May-Jun 2011;2(3):238-40. doi:10.4161/viru.2.3.16162
- Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. Journal of virology. Oct 2010;84(19):9760-74. doi:10.1128/JVI.01051-10
- Aguirre S, Maestre AM, Pagni S, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS pathogens. 2012;8(10):e1002934. doi:10.1371/journal.ppat.1002934
- Dalrymple NA, Cimica V, Mackow ER. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. MBio. May 12 2015;6(3):e00553-15. doi:10.1128/mBio.00553-15
- Miorin L, Romero-Brey I, Maiuri P, et al. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. Journal of virology. Jun 2013;87(11):6469-81. doi:10.1128/JVI.03456-12
- Fares M, Cochet-Bernoin M, Gonzalez G, et al. Pathological modeling of TBEV infection reveals differential innate immune responses in human neurons and astrocytes that correlate with their susceptibility to infection. J Neuroinflammation. Mar 3 2020;17(1):76. doi:10.1186/s12974-020-01756-x
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nature reviews. Jul 2008;8(7):559-68.
- Chmielewska AM, Gómez-Herranz M, Gach P, et al. The Role of IFITM Proteins in Tick-Borne Encephalitis Virus Infection. Journal of virology. Jan 12 2022;96(1):e0113021. doi:10.1128/jvi.01130-21
- Taylor RT, Lubick KJ, Robertson SJ, et al. TRIM79α, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe. 2011;10(3):185-196. doi:10.1016/j.chom.2011.08.004
- Upadhyay AS, Vonderstein K, Pichlmair A, et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cellular microbiology. Jun 2014;16(6):834-48. doi:10.1111/cmi.12241
- Upadhyay AS, Stehling O, Panayiotou C, Rosser R, Lill R, Overby AK. Cellular requirements for iron-sulfur cluster insertion into the antiviral radical SAM protein viperin. The Journal of biological chemistry. Aug 18 2017;292(33):13879-13889. doi:10.1074/jbc.M117.780122
- Lindqvist R, Overby AK. The Role of Viperin in Antiflavivirus Responses. DNA Cell Biol. Sep 2018;37(9):725-730. doi:10.1089/dna.2018.4328
- Lindqvist R, Upadhyay A, Overby AK. Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses. Jun 21 2018;10(7). doi:10.3390/v10070340
- Panayiotou C, Lindqvist R, Kurhade C, et al. Viperin restricts Zika virus and tick-borne encephalitis virus replication by targeting NS3 for proteasomal degradation. Journal of virology. Jan 10 2018. doi:10.1128/JVI.02054-17
- Vonderstein K, Nilsson E, Hubel P, et al. Viperin targets flavivirus virulence by inducing assembly of non-infectious capsid particles. Journal of virology. Oct 18 2017;92(1). doi:10.1128/JVI.01751-17
- Claude A, Zhao BP, Kuziemsky CE, et al. GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. The Journal of cell biology. Jul 12 1999;146(1):71-84.
- Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell. Mar 2005;16(3):1213-22. doi:10.1091/mbc.E04-07-0599
- Carpp LN, Rogers RS, Moritz RL, Aitchison JD. Quantitative proteomic analysis of host-virus interactions reveals a role for Golgi brefeldin A resistance factor 1 (GBF1) in dengue infection. Mol Cell Proteomics. Nov 2014;13(11):2836-54. doi:10.1074/mcp.M114.038984
- Lanke KH, van der Schaar HM, Belov GA, et al. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. Journal of virology. Nov 2009;83(22):11940-9. doi:10.1128/JVI.01244-09
- Liang W, Zheng M, Bao C, Zhang Y. CSFV proliferation is associated with GBF1 and Rab2. J Biosci. Mar 2017;42(1):43-56.
- Zhang N, Zhang L. Key components of COPI and COPII machineries are required for chikungunya virus replication. Biochemical and biophysical research communications. Nov 25 2017;493(3):1190-1196. doi:10.1016/j.bbrc.2017.09.142
- Fortova A, Barkhash AV, Pychova M, et al. Genetic polymorphisms in innate immunity genes influence predisposition to tick-borne encephalitis. J Neurovirol. Dec 2023;29(6):699-705. doi:10.1007/s13365-023-01182-8
- Barkhash AV, Babenko VN, Kobzev VF, Romashchenko AG, Voevoda MI. Polymorphism in the human 2′-5′-oligoadenylate synthetase genes (OAS), associated with predisposition to severe forms of tick-borne encephalitis, in populations from North Eurasia. Mol Biol (Mosk). Nov-Dec 2010;44(6):985-93.
- Barkhash AV, Perelygin AA, Babenko VN, et al. Variability in the 2′-5′-oligoadenylate synthetase gene cluster is associated with human predisposition to tick-borne encephalitis virus-induced disease. The Journal of infectious diseases. Dec 15 2010;202(12):1813-8. doi:10.1086/657418
- Lubick KJ, Robertson SJ, McNally KL, et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell host & microbe. Jul 8 2015;18(1):61-74. doi:10.1016/j.chom.2015.06.007
- Best SM, Morris KL, Shannon JG, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. Journal of virology. Oct 2005;79(20):12828-39.
- Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cellular microbiology. Mar 2008;10(3):696-712.
- Yang Q, You J, Zhou Y, et al. Tick-borne encephalitis virus NS4A ubiquitination antagonizes type I interferon-stimulated STAT1/2 signalling pathway. Emerging microbes & infections. Dec 2020;9(1):714-726. doi:10.1080/22221751.2020.1745094
- Pierson TC, Fremont DH, Kuhn RJ, Diamond MS. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell host & microbe. Sep 11 2008;4(3):229-38. doi:10.1016/j.chom.2008.08.004
- Kapadia RK, Staples JE, Gill CM, et al. Severe Arboviral Neuroinvasive Disease in Patients on Rituximab Therapy: A Review. Clin Infect Dis. Mar 21 2023;76(6):1142-1148. doi:10.1093/cid/ciac766
- Agudelo M, Palus M, Keeffe JR, et al. Broad and potent neutralizing human antibodies to tick-borne flaviviruses protect mice from disease. The Journal of experimental medicine. May 3 2021;218(5. )doi:10.1084/jem.20210236
- Kreil TR, Eibl MM. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model. Journal of virology. Apr 1997;71(4):2921-7.
- Heinz FX, Berger R, Tuma W, Kunz C. A topological and functional model of epitopes on the structural glycoprotein of tick-borne encephalitis virus defined by monoclonal antibodies. Virology. Apr 30 1983;126(2):525-37.
- Niedrig M, Klockmann U, Lang W, et al. Monoclonal antibodies directed against tick-borne encephalitis virus with neutralizing activity in vivo. Acta virologica. Jun 1994;38(3):141-9.
- Phillpotts RJ, Stephenson JR, Porterfield JS. Passive immunization of mice with monoclonal antibodies raised against tick-borne encephalitis virus. Brief report. Archives of virology. 1987;93(3-4):295-301.
- Kreil TR, Maier E, Fraiss S, Eibl MM. Neutralizing antibodies protect against lethal flavivirus challenge but allow for the development of active humoral immunity to a nonstructural virus protein. Journal of virology. Apr 1998;72(4):3076-81.
- Votiakov VI, Protas, II, Bortkevich VS, Nedz’ved MK. [Experimental study of the pathogenesis of tick-borne encephalitis]. Vopr Virusol. May-Jun 1975;(3):313-7. Eksperimental’noe izuchenie patogeneza kleshchevogo éntsefalita.
- Zlontnik I, Grant DP, Carter GB. Experimental infection of monkeys with viruses of the tick-borne encephalitis complex: degenerative cerebellar lesions following inapparent forms of the disease or recovery from clinical encephalitis. Br J Exp Pathol. Apr 1976;57(2):200-10.
- Pripuzova NS, Gmyl LV, Romanova L, et al. Exploring of primate models of tick-borne flaviviruses infection for evaluation of vaccines and drugs efficacy. PLoS One. 2013;8(4):e61094. doi:10.1371/journal.pone.0061094
- Fokina GI, Malenko GV, Levina LS, et al. Persistence of tick-borne encephalitis virus in monkeys. V. Virus localization after subcutaneous inoculation. Acta virologica. Sep 1982;26(5):369-75.
- Frolova MP, Pogodina VV. Persistence of tick-borne encephalitis virus in monkeys. VI. Pathomorphology of chronic infection in central nervous system. Acta virologica. May 1984;28(3):232-9.
- Gritsun TS, Frolova TV, Zhankov AI, et al. Characterization of a siberian virus isolated from a patient with progressive chronic tick-borne encephalitis. Journal of virology. Jan 2003;77(1):25-36.
- Kozuch O, Grulich I, Nosek J. Experimental infection of the mole with tick-borne encephalitis virus. J Hyg Epidemiol Microbiol Immunol. 1966;10(1):120-4.
- Mandl CW. Steps of the tick-borne encephalitis virus replication cycle that affect neuropathogenesis. Virus research. Aug 2005;111(2):161-74. doi:10.1016/j.virusres.2005.04.007
- Palus M, Vojtiskova J, Salat J, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. 2013;10:77. doi:10.1186/1742-2094-10-77
- Růzek D, Gritsun TS, Forrester NL, et al. Mutations in the NS2B and NS3 genes affect mouse neuroinvasiveness of a Western European field strain of tick-borne encephalitis virus. Virology. May 10 2008;374(2):249-55. doi:10.1016/j.virol.2008.01.010
- Ruzek D, Salat J, Palus M, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. Feb 5 2009;384(1):1-6.
- Engel AR, Rumyantsev AA, Maximova OA, et al. The neurovirulence and neuroinvasiveness of chimeric tick-borne encephalitis/dengue virus can be attenuated by introducing defined mutations into the envelope and NS5 protein genes and the 3′ non-coding region of the genome. Virology. Sep 15 2010;405(1):243-52. doi:10.1016/j.virol.2010.06.014
- Dobler G, Bestehorn M, Antwerpen M, Overby-Wernstedt A. Complete Genome Sequence of a Low-Virulence Tick-Borne Encephalitis Virus Strain. Genome Announc. Oct 20 2016;4(5). doi:10.1128/genomeA.01145-16
- Asghar N, Lee YP, Nilsson E, et al. The role of the poly(A) tract in the replication and virulence of tick-borne encephalitis virus. Sci Rep. Dec 16 2016;6:39265. doi:10.1038/srep39265
- Sakai M, Yoshii K, Sunden Y, Yokozawa K, Hirano M, Kariwa H. Variable region of the 3′ UTR is a critical virulence factor in the Far-Eastern subtype of tick-borne encephalitis virus in a mouse model. The Journal of general virology. Apr 2014;95(Pt 4):823-35. doi:10.1099/vir.0.060046-0
- Yoshii K, Sunden Y, Yokozawa K, et al. A critical determinant of neurological disease associated with highly pathogenic tick-borne flavivirus in mice. Journal of virology. May 2014;88(10):5406-20. doi:10.1128/jvi.00421-14
- Lindqvist R, Rosendal E, Weber E, et al. The envelope protein of tick-borne encephalitis virus influence neuron entry, pathogenicity and vaccine protection. J Neuroinflammation. Sep 28 2020;17:284. doi:10.1186/s12974-020-01943-w
- Mandl CW, Allison SL, Holzmann H, Meixner T, Heinz FX. Attenuation of tick-borne encephalitis virus by structure-based site-specific mutagenesis of a putative flavivirus receptor binding site. Journal of virology. Oct 2000;74(20):9601-9.
- Mandl CW, Holzmann H, Meixner T, et al. Spontaneous and engineered deletions in the 3′ noncoding region of tick-borne encephalitis virus: construction of highly attenuated mutants of a flavivirus. Journal of virology. Mar 1998;72(3):2132-40.
- Michelitsch A, Fast C, Sick F, et al. Long-term presence of tick-borne encephalitis virus in experimentally infected bank voles (Myodes glareolus). Ticks Tick Borne Dis. Jul 2021;12(4):101693. doi:10.1016/j.ttbdis.2021.101693
- Chiffi G, Grandgirard D, Stöckli S, Valente LG, Adamantidis A, Leib SL. Tick-borne encephalitis affects sleep-wake behavior and locomotion in infant rats. Cell Biosci. Aug 2 2022;12(1):121. doi:10.1186/s13578-022-00859-7
- Scroggs SLP, Offerdahl DK, Stewart PE, Shaia C, Griffin AJ, Bloom ME. Of Murines and Humans: Modeling Persistent Powassan Disease in C57BL/6 Mice. mBio. Apr 25 2023;14(2):e0360622. doi:10.1128/mbio.03606-22
- Mlera L, Meade-White K, Saturday G, Scott D, Bloom ME. Modeling Powassan virus infection in Peromyscus leucopus, a natural host. PLoS neglected tropical diseases. Jan 2017;11(1):e0005346. doi:10.1371/journal.pntd.0005346
- Pogodina VV, Frolova TV, Frolova MP, Sobolev SG, Shamanin VA, Pletnev AG. Molecular hybridization with cloned fragments of tick-borne encephalitis (TBE) virus cDNA in acute and chronic TBE infection. Acta virologica. Jan 1991;35(1):71-80.
- Frolova TV, Pogodina VV, Frolova MP, Karmysheva V. [Characteristics of long-term persisting strains of tick-borne encephalitis virus in different forms of the chronic process in animals]. Vopr Virusol. Jul-Aug 1982;27(4):473-9. Kharakteristika dlitel’no persistiruiushchikh shtammov virusa kleshchevogo éntsefalita pri ralichnykh formakh khronicheskogo protsessa u zhivotnykh.
- Andzhaparidze OG, Rozina EE, Bogomolova NN, Boriskin YS. Morphological characteristics of the infection of animals with tick-borne encephalitis virus persisting for a long time in cell cultures. Acta virologica. May 1978;22(3):218-24.
- Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. Dec 1989;1(3):285-96.
- Boyer JC, Haenni AL. Infectious transcripts and cDNA clones of RNA viruses. Virology. Feb 1994;198(2):415-26. doi:10.1006/viro.1994.1053
- Aubry F, Nougairede A, Gould EA, de Lamballerie X. Flavivirus reverse genetic systems, construction techniques and applications: a historical perspective. Antiviral Res. Feb 2015;114:67-85. doi:10.1016/j.antiviral.2014.12.007
- Mandl CW, Ecker M, Holzmann H, Kunz C, Heinz FX. Infectious cDNA clones of tick-borne encephalitis virus European subtype prototypic strain Neudoerfl and high virulence strain Hypr. The Journal of general virology. May 1997;78 ( Pt 5):1049-57. doi:10.1099/0022-1317-78-5-1049
- Gritsun TS, Gould EA. Infectious transcripts of tick-borne encephalitis virus, generated in days by RT-PCR. Virology. Dec 20 1995;214(2):611-8. doi:10.1006/viro.1995.0072
- Gritsun TS, Gould EA. Development and analysis of a tick-borne encephalitis virus infectious clone using a novel and rapid strategy. Journal of virological methods. Dec 1998;76(1-2):109-20.
- Aubry F, Nougairede A, de Fabritus L, Querat G, Gould EA, de Lamballerie X. Single-stranded positive-sense RNA viruses generated in days using infectious subgenomic amplicons. The Journal of general virology. Nov 2014;95(Pt 11):2462-7. doi:10.1099/vir.0.068023-0
- Yoshii K, Konno A, Goto A, et al. Single point mutation in tick-borne encephalitis virus prM protein induces a reduction of virus particle secretion. The Journal of general virology. Oct 2004;85(Pt 10):3049-58. doi:10.1099/vir.0.80169-0
- Rumyantsev AA, Murphy BR, Pletnev AG. A tick-borne Langat virus mutant that is temperature sensitive and host range restricted in neuroblastoma cells and lacks neuroinvasiveness for immunodeficient mice. Journal of virology. Feb 2006;80(3):1427-39. doi:10.1128/JVI.80.3.1427-1439.2006
- Kofler RM, Hoenninger VM, Thurner C, Mandl CW. Functional analysis of the tick-borne encephalitis virus cyclization elements indicates major differences between mosquito-borne and tick-borne flaviviruses. Journal of virology. Apr 2006;80(8):4099-113.
- Markoff L. 5′- and 3′-noncoding regions in flavivirus RNA. Advances in virus research. 2003;59:177-228.
- Silva PA, Pereira CF, Dalebout TJ, Spaan WJ, Bredenbeek PJ. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. Journal of virology. Nov 2010;84(21):11395-406. doi:10.1128/JVI.01047-10
- Funk A, Truong K, Nagasaki T, et al. RNA structures required for production of subgenomic flavivirus RNA. Journal of virology. Nov 2010;84(21):11407-17. doi:10.1128/JVI.01159-10
- Lin KC, Chang HL, Chang RY. Accumulation of a 3′-terminal genome fragment in Japanese encephalitis virus-infected mammalian and mosquito cells. Journal of virology. May 2004;78(10):5133-8.
- Pijlman GP, Funk A, Kondratieva N, et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell host & microbe. Dec 11 2008;4(6):579-91. doi:10.1016/j.chom.2008.10.007
- Roby JA, Pijlman GP, Wilusz J, Khromykh AA. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses. Feb 2014;6(2):404-27. doi:10.3390/v6020404
- Schnettler E, Tykalova H, Watson M, et al. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res. Aug 2014;42(14):9436-46. doi:10.1093/nar/gku657
- Gritsun TS, Venugopal K, Zanotto PM, et al. Complete sequence of two tick-borne flaviviruses isolated from Siberia and the UK: analysis and significance of the 5′ and 3′-UTRs. Virus research. May 1997;49(1):27-39.
- Hoenninger VM, Rouha H, Orlinger KK, et al. Analysis of the effects of alterations in the tick-borne encephalitis virus 3′-noncoding region on translation and RNA replication using reporter replicons. Virology. Aug 1 2008;377(2):419-30. doi:10.1016/j.virol.2008.04.035
- Sakai M, Muto M, Hirano M, Kariwa H, Yoshii K. Virulence of tick-borne encephalitis virus is associated with intact conformational viral RNA structures in the variable region of the 3′-UTR. Virus research. May 4 2015;203:36-40. doi:10.1016/j.virusres.2015.03.006
- Asghar N, Lindblom P, Melik W, et al. Tick-borne encephalitis virus sequenced directly from questing and blood-feeding ticks reveals quasispecies variance. PLoS One. 2014;9(7):e103264. doi:10.1371/journal.pone.0103264