Chapter 7:
Immunology of TBEV infection
Kyra D. Zens and Rahel Ackermann-Gäumann
Key points
- The host immune response to Tickborne Encephalitis Virus (TBEV) infection involves the coordination of multiple immune subsets at several distinct tissue sites over time.
- Contributions from both early innate and later adaptive immune responses are critical in controlling TBEV infection.
- Early innate immune responses are driven by Type I interferon-mediated signaling and are dominated by neutrophils and natural killer cells.
- Antibody-mediated humoral responses and T cell-mediated cellular immune responses both contribute to adaptive immune control of TBEV infection.
- The mechanisms of Central Nervous System (CNS) pathogenesis during Tickborne Encephalitis (TBE) remain unclear but may involve a combination of direct viral cytopathic effects and immune-mediated damage.
- An improved understanding of host immune responses during TBE could aid in the development of improved therapies.
Introduction
Tick-borne Encephalitis (TBE) is a severe, vaccine-preventable disease of the Central Nervous System (CNS) caused by the tick-borne encephalitis virus (TBEV). The virus is primarily transmitted to humans through the bite of infected Ixodid ticks, though an estimated 1% of cases occur via alimentary transmission1,2 and rare cases of transmission through organ or blood donation have been documented.3,4 An estimated 70% of TBEV exposures are asymptomatic.5-7 The remaining 30% of individuals experience a brief, asymptomatic incubation phase,1,2,8 followed by a period of viremia accompanied by febrile, influenza-like illness. While most individuals recover without further symptoms, approximately 30% progress to a second phase of illness characterized by CNS involvement.1,2,8,9 While some individuals transition directly from the first systemic phase to the second CNS phase, referred to as “monophasic” disease, most experience a short symptom-free interval of approximately 1 week between these two phases, which is referred to as “biphasic” disease. Factors driving a monophasic versus biphasic disease course are not completely clear. Data clearly linking viral subtype to clinical disease course are lacking, though it is believed that monophasic disease, as well as a more severe disease course, are more common after infection with the Siberian (TBEV-Sib) and Far Eastern (TBEV-FE) viral subtypes compared to the European (TBEV-Eu) subtype.1,10 Differences in virulence factors responsible for distinct pathologies between viral subtypes, however, have yet to be described and confounding factors, such as age, chronic conditions, or possibly even regional differences in medical practices could further play roles.
The immune responses which protect individuals against disease represent a complex interplay between many distinct cell types at various times and over different locations. Innate immunity comprises the “first line” defenses following pathogen exposure, acting broadly within the first hours to days following infection to protect against invaders. TBEV belongs to the genus Orthoflavivirus, which also includes the clinically-relevant, arthropod-borne viruses Dengue, West Nile, Yellow Fever, Japanese Encephalitis, and Zika1,2,11 and early immune responses to TBEV infection share many features with these viruses.12 Adaptive immune responses, comprised by both humoral (i.e. antibody), and cell-mediated (i.e. T cell) responses, take more time to be established, on the order of days to weeks, as they require the initial activation of the innate immune system. Adaptive immunity, however, provides highly-specific protection against invading pathogens, and further offers immune memory – a subset of cells which are maintained long-term (up to decades), and provide rapid protection upon later re-exposure to the same pathogen.
In this chapter, we summarize the early innate and adaptive immune responses to TBEV infection as well as discuss potential mediators of long-term immune memory protective against later viral reinfection.
TBEV transmission and early local innate immune responses
Skin is perhaps the most important immune organ in that it acts as an initial physical barrier to many infectious organisms. The skin further contains many specialized immune cells, including resident dendritic cell (DC) subsets, natural killer (NK) cells, and T cell subsets, among others (Figures 1, 2). Transmission of TBEV through tick bites helps the virus to partially circumvent skin’s role as a protective physical barrier. Furthermore, factors present within the tick’s saliva, including various compounds which help to suppress local innate responses as well as the initiation of adaptive immunity,13-15 further facilitate viral transmission.
Figure 1: TBEV transmission and timeline of viral and host immune response
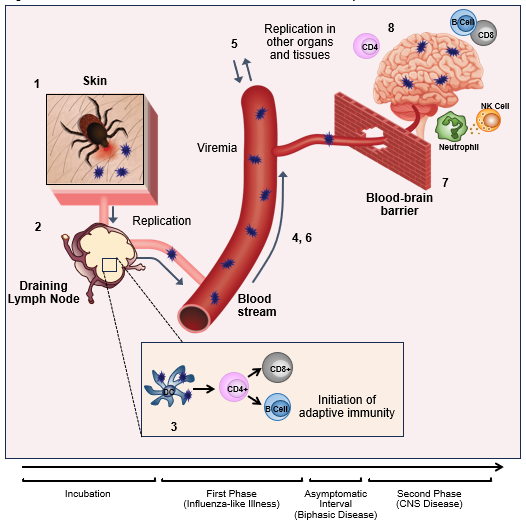
Click the image above to enlarge
1) TBEV is transmitted by the bite of an infected tick. 2) The virus infects dendritic cells (DCs) within the kin which traffic to the draining lymph node where the virus replicates further. 3) Presentation of TBEV-derived antigens by infected DCs results in the activation of adaptive immune responses; these take, however weeks to fully develop. 4) The virus is able to spread from the draining lymph node into the blood; during this primary viremia, the host experiences the first symptomatic phase of illness. 5) During primary viremia the virus seeds peripheral organs and replicates further within the tissues. This leads to 6) a second period of virema during which the virus is able to 7) cross the blood brain barrier (BBB). 8) Involvement of the CNS leads to the second phase of disease (in individuals experiencing biphasic illness), neutrophils, T cells, NK cells and B cells can be detected in the CNS.
The innate immune system is the first line of defense against infection and is especially crucial for so-called “naïve” hosts that have not yet encountered a specific pathogen and developed corresponding adaptive immune memory. Following exposure to TBEV-infected ticks, local skin inflammatory responses begin within 1-3 hours of attachment.16-18 Pathogen recognition by the innate immune system depends on the host’s expression of pattern recognition receptors (PRRs), which identify conserved moieties expressed by invading microorganisms. Toll-Like Receptors (TLRs) and Retinoic Acid-Inducible Gene I (RIG-I)-Like Receptors (RLRs), including RIG-I and Melanoma Differentiation-Associated protein 5 (MDA5), are important in the detection of RNA viruses. Upon activation in this context, PRRs initiate signaling cascades that activate the Interferon (IFN) regulatory factor 3 (IRF-3) signaling pathway, leading to the production of IFN. The role of TLR signaling in protecting against TBEV infection is not well-defined, although TLR-3 and possibly TLR-7, may be involved.19,20 Roles for RIG-I and MDA5 in the innate immune recognition of TBEV proteins, including non-structural protein 5 (NS5) have been demonstrated.17 This recognition leads to an early immune response dominated by type I IFN (IFN-a and IFN-b), which seems to be the key mediator of protection during early infection in both in vitro and in vivo models.21,22 In line with this, mice that lack the IFN-α/β receptor (IFNAR) are unable to control TBEV infection and studies of polymorphisms in innate immune response genes in patients have identified variations in the interferon-induced antiviral proteins oligoadenylate synthetase 2 (OAS2) and 3 (OAS3), which may predispose individuals to the development of clinical TBE.23 While it has been established that differing strains of TBEV can elicit distinct symptoms in mouse models of disease20,24 the immunological mechanisms underlying these differences remain incompletely described, though early differences in innate responses due to viral evasion could potentially play an important role.
Local dendritic cell (DC) responses
DCs represent a group of cells with a range of functions including acting as a major source of type I IFN during viral infection and playing critical roles in antigen presentation and the activation of adaptive immune responses (Figure 2, 3). DCs are often described as the interface between the innate and adaptive immune systems. After TBEV is transmitted, skin-localized DCs are among the first cell types to be infected and they likely play an important role in viral trafficking. In addition, infection of DCs in vitro with Langat virus (LGTV), an attenuated member of the TBE serogroup, has been shown to inhibit type I IFN signaling and reduce IL-12 production – an activator of type 1 adaptive immune responses which are crucial in controlling viral infections.25 Inhibition of DC type I IFN signaling by the virus, therefore, acts as an important host evasion mechanism and helps to suppress the ensuing immune response. Interestingly, infection of DCs with distinct TBEV strains in vitro has been demonstrated to result in distinct functional capacities, also impacting later activation of CD4+ T cells.20 In addition, higher viral infectious doses in mice result in delayed DC activation and IFN production, and may impact viral spread to the CNS.20
Figure 2: TBEV transmission and initiation of host immune responses
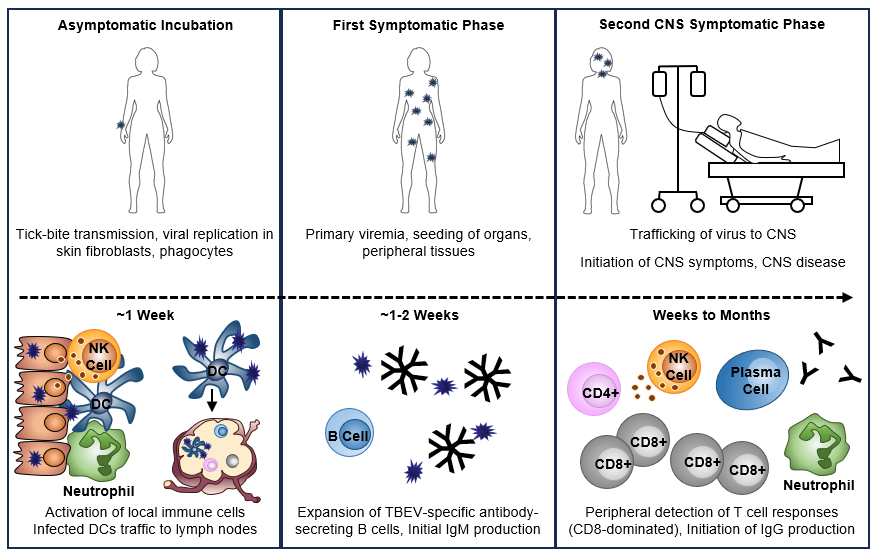
Click the image above to enlarge
Following tick bite-mediated transmission of TBEV, the virus first infects local skin cells including fibroblasts and phagocytic cells. This leads to the rapid initiation of innate immune responses resulting in the recruitment of additional immune cells to the bite site. Infected DCs are thought to migrate to the draining lymph nodes where they begin to initiate TBEV-specific adaptive immune responses. The virus next disseminates to the organs and peripheral tissues. During this primary viremia, the host experiences the first symptomatic phase of illness. As IgM and antibody-secreting B cells can be detected in patients with biphasic illness upon hospitalization indicates that these responses likely begin during the first phase of illness or short recovery period prior to initiation of CNS symptoms. It is not yet known at what point during the process of viral dissemination that TBEV reaches the CNS. In individuals experiencing biphasic illness with CNS involvement, neutrophils, T cells, NK cells and B cells can be detected in the CNS. Virus-specific T cells and activated NK cells can also be found in peripheral blood. T cell responses, which are strongly CD8-biased, are detected in the blood and peak approximately 1 week after CNS symptom onset. Both anti-TBEV IgM and IgG antibodies are detected in serum during the second phase of TBE. IgM responses peak and begin to transition to IgG responses, which dominate during convalescence. While this figures depicts what is currently known for TBEV infection and the initiation of immune responses during TBE disease, the complete mechanism for this process remains to be understood.
Primary viremia and seeding of peripheral tissues
In the absence of early immune control within the skin, TBEV next traffics to the draining lymph nodes (Figures 1,2). This process is not completely understood, but likely occurs during the asymptomatic incubation phase with the migration of virally-infected phagocytes or DCs from the skin playing an important role.26 Once within the lymph nodes, the virus replicates and eventually seeds peripheral organs (Figures 1,2). During this viral expansion the host experiences a period of systemic viremia,1,2,8,27,28 which corresponds to the first symptomatic phase of disease. An estimated 70% of individuals control the infection at this stage, though the mechanisms of this control are not clear. Work in a mouse LGTV model has demonstrated a critical role for the type-I IFN response in limiting initial viral replication and systemic spread.29 This is likely important in the context of TBEV infection as well and suggests a key role for innate immunity in not only early local, but also early systemic immune control of TBEV infection. This is supported by the fact that, due to delayed initiation of adaptive immunity, antibody and T cell responses are absent in the first weeks after pathogen encounter in “naïve” hosts and would, therefore, not be expected to contribute to protection.
Secondary viremia and CNS disease
As described, the remaining 30% of individuals unable to control TBEV during the early local and systemic stages of infection progress to disease which includes CNS involvement. TBEV is neurotropic – preferentially infecting cells of the nervous system. TBEV replication, for example, has been shown to be 10,000-fold higher in human neuronal cells compared to epithelial cells.30 The ability of the virus to cross the blood brain barrier and invade the CNS is the root cause of clinical disease (Figures 1,2). In some cases, this progression can directly follow the initial febrile, influenza-like illness (monophasic disease), though most individuals experience a short symptom-free interval prior to CNS disease progression (biphasic disease). In a biphasic disease course, CNS symptoms may occur anywhere from 4 days up to more than 60 days after viral exposure.1,2,8 Differences in immune control between monophasic and biphasic illness are not well-defined but may also be driven by differences in early innate control rather than differences in later adaptive responses. A recent study comparing monophasic and biphasic disease found that patients with a biphasic disease course were younger and had fewer comorbidities. Levels of proinflammatory cytokines in the CSF were also lower in a biphasic course suggesting less severe disease.31 In either case, the route by which CNS seeding occurs is not well-understood, though breakdown of the blood brain barrier (BBB) does not appear to be necessary for TBEV entry into the brain 32,33 and the virus is no longer present in the blood once CNS involvement is clinically apparent. However, a recent study demonstrating TBEV transmission following organ transplantation brings into question whether the virus may persist in the peripheral tissues for prolonged periods following infection, perhaps even when no longer detectable in the blood.3
Much of what is known about immune responses to TBEV in humans has been studied during the CNS phase of disease as patients generally present to the clinic only after neurological symptoms have begun. Several studies have evaluated serum cytokine responses in these patients and factors including Chemokine (C-C-motif) Ligand (CCL)5, CCL7, Chemokine (C-X-C-motif) Ligand (CXCL)10, CXCL11, CXCL13, Interferon (IFN)-γ, Interleukin (IL)-1 α, IL-6, IL-15, IL-18, and Tumor Necrosis Factor (TNF)-α have been found to be upregulated, among others.34-40 A “TBE-specific” cytokine profile, however, which could be useful for diagnostic purposes, has not been defined. Importantly, the entry of immune cells into the brain, which may contribute to immunopathology observed during severe infection in animal studies,33 relies on cytokine-mediated trafficking. In TBE patients, increased levels of CCL534 and CXCL1034,37 in the cerebral spinal fluid (CSF) may be involved in T cell recruitment into the brain during disease through CCR534 and CXCR3-mediated37 trafficking. Similarly, levels of CXCL10 are increased in the sera and brains of mice during TBEV infection.41 Strong cytokine responses in the brain, coupled with very low neutralizing antibody responses, have been linked to enhanced disease and death.42 Interestingly, polymorphisms in CCR5, which is an important driver of leukocyte migration, have been implicated in TBE disease susceptibility and severity.19
Natural killer (NK) cell responses during CNS disease
NK cells (Figure 3) are a subset of cytotoxic innate lymphocytes which play important roles in eliminating virally-infected and tumor cells. While not much is known about the role of NK cells in TBE prior to the development of CNS disease, NK cell-associated cytokines, including IL-12, IL-15, IL-18, IFN-γ, and TNF-α are upregulated in patient sera43 and NK cells can further be detected in the CSF; indicating their migration to the CNS.44 Interestingly, while NK cells detected in the peripheral blood of patients have an activated (CD57+ CD56dim) phenotype,43 they appear to be poorly functional, possibly indicating limited protective capacities.43 Thus, clear roles for NK cells in the context of TBE have not yet been defined, particularly during mild disease where their function may be distinct from that observed in severe disease.
Figure 3: Innate and adaptive immune cells known to be involved in TBE disease
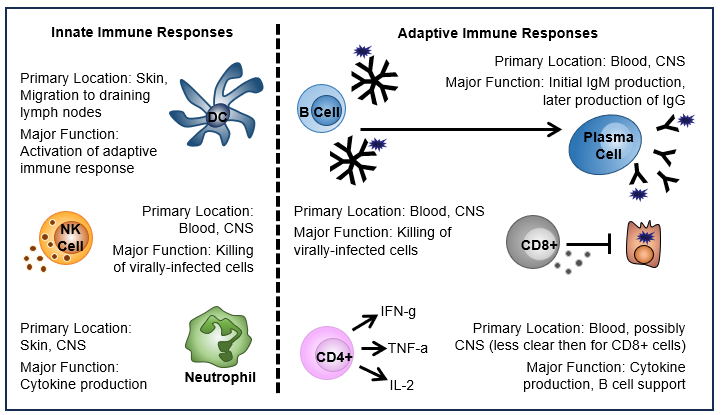
Click the image above to enlarge
The localization and function of innate and adaptive immune cell subsets described in the context of TBEV infection and TBE disease. DCs are thought to be involved in the initial trafficking of TBEV to the draining lymph nodes following infection. Their major role is in the initiation of later adaptive immune responses. NK cells can be found in the cerebral spinal fluid (CSF) of CNS disease patients and NK cells detected in the blood have an activated (CD57+ CD56dim) phenotype, but lower degranulation and expression of perforin and granzyme B suggesting reduced functionality. Neutrophils are likely among the first cell types at the site of infection and can be infected by TBEV. In CNS disease patients they are present in the CSF and may positively correlate with disease severity. B cells are a key mediator of the adaptive immune response to TBEV as the are responsible for antibody production. Initially IgM is produced, followed by IgG. T cell responses are CD8-biased, though CD4+ T cells are important in providing the B cell help necessary for antibody production.
Neutrophil responses during CNS disease
Neutrophils are a critical phagocytic cell subset during the early immune response to viral infections and are major producers of inflammatory cytokines. In tick feeding experiments, neutrophils are attracted to the bite site and can also be infected with TBEV.26 Like NK cell responses, however, little is known about their role in protection prior to CNS disease. One study found that neutrophils are universally present in the CSF of TBE patients, and, that IL-8, a neutrophil chemoattractant, is the most abundant CSF cytokine.45 In the same study, neutrophil counts positively correlated with disease severity in patients and their continued detection in CSF samples into convalescence was associated with neurologic sequelae.45 Supporting this, work in a mouse LGTV model demonstrated increased neutrophil migration into the CNS, and, further, that depletion of neutrophils reduced viral loads, decreased immunopathology, and improved survival.46 Together these findings suggest that neutrophils may play a role in immunopathology, at least in the context of severe TBE, making them a potential immunotherapeutic target.
Cellular immune responses to TBEV infection
Cellular immunity forms one arm of the so-called “adaptive” immune system (Figure 3). A key feature of adaptive immune responses is the ability to form immune memory following primary pathogen exposure, which is able to provide rapid protective responses upon later pathogen re-encounter. Cellular immunity relies primarily on T cell-mediated immune responses. While T cell responses during TBEV infection are less studied and less understood than humoral responses, T cells seem to play an important role in protection. As with early innate immune responses, a major issue in our understanding of cellular immunity during TBEV infection is that most studies are conducted in patients with relatively severe disease, and late during the disease course – namely after CNS involvement. As a consequence, our understanding of what constitutes “ideal” protective immunity is limited.
CD4+ T cells
Cytokine production is arguably the most important function of CD4+ T cells during antiviral immune responses. These cells are also essential in providing the help necessary for B cells to effectively produce antibodies. Like other orthoflaviviruses, the TBEV genome encodes seven non-structural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5),1,2,11 and three structural proteins (capsid (C), two membrane-associated proteins; precursor of membrane/membrane (prM/M), and envelope (E)).1,2,11 These structural proteins appear to be the major targets of CD4+ T cell responses during TBEV infection.47,48 In clinical TBE cases, T cell activation has been observed to peak approximately one week after hospitalization, indicating that primary T cell responses are delayed until the CNS phase of illness, at least in severe disease.49,50 Whether this is the case in mild infections is not clear.
The majority of CD4+ T cells observed during TBEV infection are polyfunctional, producing mainly IL-2, TNF-α, and IFN-γ; the major cytokines of type 1 immune responses (Figure 3).47,50 IFN-γ-mediated responses, in particular, are known to be important in the control of viral infections and are often also associated with direct antiviral effector functions in CD4+ T cells. CD4+ T cells appear to have a moderate activation phenotype during TBE infection, suggesting that they may play a less important role in direct viral clearance, but also, may have less immunopathogenic potential, than, for example, CD8+ T cells.51 In line with their potential protective roles, adoptive transfer of CD4+ T cells has been shown to protect against lethal disease in TBEV-infected Severe Combined Immunodeficiency (SCID; no T or B cells) mice.30
CD8+ T cells
CD8+ T cells, also known as cytotoxic T cells, play crucial roles in viral infection through their ability to identify and destroy infected host cells, thereby limiting viral replication and spread (Figure 3). In contrast to CD4+ T cells, which appear to target TBEV structural proteins during infection, the CD8+ T cell response appears primarily to target NS proteins; among 6 CD8+ T cell epitopes identified in one study, all were derived from NS proteins.52 In TBE patients, peak T cell responses are observed approximately 1 week following hospitalization with CD8+ T cell activation substantially increased compared to CD4+ T cells, indicating that responses tend to be CD8-dominated.51 These CD8+ T cells further displayed an effector phenotype (CD45RA-CCR7),51,52 and had a highly-activated Eomes+Ki67+T-bet+ transcriptional profile.51 As patients became convalescent, virus-specific CD8+ T cells transitioned to an Eomes-Ki67-T-bet+ phenotype,51 consistent with a type 1 effector memory (TEM) population.
While immune responses during acute CNS disease are CD8-dominated (Figure 2), the role of these CD8+ T cells in immunopathology versus protection during TBE disease is unclear. Results in animal studies have also been mixed. CCR5-deficient animals experienced a temporal lag in lymphocyte migration into the CNS during LGTV infection which resulted in increased mortality. This was, however, alleviated by adoptive transfer of wildtype (but not CCR5-deficient) T cells, demonstrating the importance to T cell responses in protection from lethal infection.46 In contrast, survival following lethal TBEV infection in SCID and CD8-knockout mice was increased compared to wildtype or mice with adoptively transferred CD8+ T cells, demonstrating that CD8+ T cells can also contribute to lethal infection.30 Similarly, CD8+ T cell infiltrates are commonly found in the post-mortem brains of fatal TBE cases,53-55 and a separate study found that, in severely infected patients, nearly all virus-specific CD8+ T cells expressed a4 and b1 integrins (VLA-4), which are important in lymphocyte homing and can mediate trafficking across the BBB.52 However, breakdown of the BBB during infection in mice was observed in both wildtype and CD8-knockout animals, indicating that CD8+ T cells themselves are not responsible for BBB permeability during disease.33 Interestingly, in a mouse model of TBEV infection, TCR CDR3 gene usage differed between lethally and non-lethally infected mice, although no differences in T-cell activation markers or apoptosis-related genes were observed, suggesting that disease severity may be related to antigen specificity, rather than simply the number or activation level of brain-infiltrating T cells.56 While the mechanism by which TBEV causes CNS destruction remains unclear, a combination of both direct neuronal damage by the virus and indirect damage caused by the immune response may be involved.
Humoral immune responses in TBEV infection
Humoral immunity, mediated by antibodies produced by B cells, is the arm of the adaptive immune response which acts to neutralize and eliminate extracellular microbes and microbial toxins. The humoral immune response plays a critical role in protecting the host from viral infections with antibodies neutralizing virus binding and entry to host cells, as well as coating viral particles to induce their uptake and destruction by phagocytic immune cells; a process termed opsonization. The long-term maintenance of memory B cells enables the immune system to respond more quickly and effectively upon reinfection as these cells rapidly differentiate into antibody-producing plasma cells when they encounter the same pathogen again; in the case of TBEV, helping to eliminate the virus before it can cause widespread infection and disease. Humoral immunity likely plays a crucial role in preventing TBE by generating antibodies that specifically target TBEV. These antibodies neutralize the virus and prevent its spread, helping to limit infection severity and, also, by providing long-term immunity against future viral exposure (Figure 3).
B cells
In contrast to T cells, which, as discussed, peak in their response approximately 1 week post-symptomatic CNS disease, TBEV-specific humoral responses are observed even earlier on during infection (Figure 1). Among TBE patients, activated antibody-producing B cells are already detected at the time of hospital admission. Furthermore, these cells do not appear to expand at this point in time, indicating that these responses are likely initiated prior to CNS-symptomatic disease, perhaps following initial viremia during the asymptomatic interval before CNS symptoms appear.57 Similarly, in the same study, all patients presented with detectable TBEV-specific IgM and IgG antibodies upon admission which were maintained into convalescence.57 In comparing immune responses in the peripheral blood and CNS during TBEV infection, several studies have suggested that type 1 cellular immune responses tend to be higher in the CSF, 36,38,44,58 while Th17-type responses, dominated by follicular helper T cells which provide help to antibody-producing B cells, and B cell responses are more pronounced in the blood.36,38,44,58 Together, these findings indicate that B cells and antibody-mediated responses are likely important in controlling the viremic stages of infection where TBEV may spread and seed several peripheral tissues.
Antibody responses
The dynamics of antibody responses following TBEV infection and primary vaccination have been well-reviewed9,10 and humoral immunity is better understood than cellular immunity. While anti-TBEV antibodies are not yet present during the initial viremic phase of TBEV infection,27,28 both IgM, and later on IgG, can be detected in serum during the CNS phase of illness59 consistent with a limited contribution of adaptive immunity in the early immune control of TBEV during the initial viremic stage of infection. Serum IgM begins to rise within the first six days of CNS symptoms, drops again within six weeks, but remains detectable for several months after infection.59,60 In contrast, serum IgG levels increase moderately during the CNS symptomatic phase of disease and peak much later – approximately 6 weeks after the onset of the first neurological symptoms.10,59-62 IgG responses, however, are durable, possibly persisting life-long following infection, and likely play a major role in protection from reinfection.59,63
B cell and antibody-mediated responses seem to primarily target the viral E and, to some extent, NS1 proteins. The E glycoprotein mediates viral binding and entry into host cells and is the primary target for neutralizing antibodies during infection as well as in response to TBE vaccination.64 More than 12 distinct epitopes within E have been identified which elicit antibodies characterized by varying degrees of neutralization potency.64 In contrast, NS-specific antibodies do not directly neutralize virus infectivity, but likely protect via other mechanisms64 and several studies have shown that NS1-specific antibodies help to protect against TBE.65-71 Assessment of anti-NS1 antibody titers may help to distinguish between TBEV infection and previous TBE vaccination, important during vaccine breakthrough infections, as NS proteins are produced mainly during viral replication.72-74 Low levels of NS1-specific antibodies, however, may also be generated in response to vaccination.75
Antibody neutralization potential
Neutralizing antibodies are widely considered to be a key mediator of protective immunity against TBE, and, indeed, neutralizing titers of 1:10 or greater are considered a surrogate measurement for the “correlate of protection” against TBE.76,77 Orthoflaviviral neutralizing antibodies have been shown to interfere with the process of virus-induced membrane fusion, preventing infection of target host cells.78-80 Other mechanisms of action have been suggested to include blocking the binding of the viral particles to cellular receptors, blocking the interaction of the virion with cellular receptors through steric hindrance, or blocking membrane fusion inside endosomes or phagosomes within the host cells through the cross-linking of E molecules.81 Importantly, though, orthoflavivirus neutralization appears to be a “multiple hit” phenomenon requiring engagement by more than a single antibody.64 It is plausible that the mechanism of neutralization of many E-specific antibodies involves both steps of virus entry and is modulated by the composition of antibody populations in polyclonal sera.82
Epitopes involved in TBEV neutralization have been mapped to each of the three viral E protein domains, to domain-overlapping sites within a single E protein monomer, to E protein dimer-specific sites, and to E protein sites requiring the quaternary arrangement found only within viral particles.82 The dominance of antibodies to different E domains appears to be heavily impacted by host-species-specific, as well as virus-specific, factors. Many of the most potent orthoflaviviral neutralizing antibodies characterized to date recognize the upper lateral surface of domain III of the E protein (EDIII) that protrudes from the surface of the virion; however these antibodies are major contributors to the neutralizing responses observed in mice but not in humans.64,83 In contrast, antibodies against domains I and II, EDI and EDII, dominate the human immune response to TBEV.84 Due to the potent neutralizing activity of anti-EDIII antibodies, though, vaccination or therapeutic strategies focusing on this domain could be beneficial.78
Cross-neutralization between Orthoflaviviruses
While available TBE vaccines designed to protect against the TBEV-Eu subtype have been shown to additionally protect against TBEV-Sib and TBEV-FE subtypes,85-87 antigenic similarities between orthoflaviviruses can also lead to the generation of both species-specific, as well as orthoflavivirus cross-reactive antibodies in response to infection.88 For instance, a study has demonstrated that individuals who had received vaccinations against Japanese Encephalitis virus, Yellow Fever virus, and TBEV were able to neutralize Louping-ill virus and to a lesser degree West Nile virus and Dengue virus.89 Similarly, TBEV neutralizing antibodies have been shown to be broadly active against other tick-borne orthoflaviviruses including Louping ill virus, Langat virus, and Omsk Hemorrhagic Fever virus,78 and the immune response generated following TBEV vaccination can protect against Omsk Hemorrhagic Fever virus, Kyasanur Forest Disease virus and Alkhumra virus.90,91 However, cross-neutralizing antibodies are usually not durable and cross-neutralization is retained only a few months.92 And while cross-neutralization might provide a certain level of cross-protection from infection, such pre-existing immunity to other orthoflaviviruses may also impair or modulate the immune response to TBEV vaccination. For instance, in a cross-sectional study examining risk factors for seronegativity despite vaccination, individuals being vaccinated against Yellow Fever or Japanese Encephalitis virus were less likely to be seropositive for neutralizing TBEV antibodies.93 Similarly, both an increase in broadly orthoflavivirus cross-reactive antibodies and an impairment in TBEV-neutralizing activity in individuals with previous vaccination against Yellow Fever virus have been demonstrated.94 Interestingly, broadly cross-reactive antibodies are more frequently observed in individuals post-vaccination than post-infection.84 On a molecular basis, cross-reactive antibodies are specific for a cluster of epitopes that are partially occluded in the cage-like assembly of E proteins at the surfaces of infectious virions and involve—but are not restricted to—amino acids of the highly conserved internal fusion peptide loop. The cryptic properties of these sites can provide an explanation for the observed low neutralizing potency of broadly cross-reactive antibodies, despite their specificity for a functionally important structural element in the E protein.88,95-97
Durability of protection
Following TBEV infection antibody titers remain stable at high levels over many years.98,99 Titers following infection are also comparable between both older and younger individuals,98,99 in contrast to vaccination where titers tend to be inversely correlated with age. While it is thought that IgG generated in response to infection may possibly persist lifelong, providing continued protection from reinfection,10 a comparison of seroprevalence and average TBE incidence rates from the 1980s through 2001 suggests that this might not be the case.100 These results suggest that, in order to err on the side of caution, additional booster vaccinations should be considered, even for recovered TBE patients. However, more evidence is necessary to better understand the duration of immunity following TBEV infection to help define best practices for vaccination and ensure continued protection.
Conclusion
TBE is a complex disease which requires the host to respond to viral infection at several distinct tissue sites over a prolonged period of time. Despite considerable insights into innate and adaptive immunity against TBEV infection, numerous questions remain. Early in infection, for example, the immune response is critically shaped by local responses within the skin. Determining whether local trained innate immune responses or “tissue-resident” T or B cell subsets could protect from TBEV infection, providing rapid control at the initial infection site before viral spread, is an interesting area worth further exploration. Furthermore, understanding and identifying specific cytokine expression profiles contributing either to protection or immunopathology, early in acute TBE disease holds therapeutic promise. In terms of adaptive immunity, while antibody responses have been extensively studied in TBE disease, memory B and T cell responses may also act as important mediators of protection. Additional research focusing on the functions of these adaptive immune subsets, particularly in asymptomatic and mild cases, is crucial to defining “ideal” protective immune responses and establishing a baseline for vaccine-mediated immunity. Ultimately, though, a better understanding of the immune responses involved in protection and possibly also immunopathology of TBE can help in the development of effective strategies for its prevention, diagnosis, and treatment.
Contact
Kyra D. Zens
zens@immunology.uzh.ch
Authors
Kyra D. Zens and Rahel Ackermann-Gäumann
Citation
Zens KD. Ackermann-Gäumann R. Immunology of TBEV Infection. Chapter 7. In: Dobler G, Erber W, Bröker M, Chitimia-Dobler L, Schmitt HJS, eds. The TBE Book. 7th ed. Singapore: Global Health Press; 2024. doi:10.33442/26613980_7-7
References
- Lindquist L, Vapalahti O. Tick-borne encephalitis. Review. Lancet. May 31 2008;371(9627):1861-71. doi:10.1016/s0140-6736(08)60800-4
- Gritsun TS, Lashkevich VA, Gould EA. Tick-borne encephalitis. Review. Antiviral Res. Jan 2003;57(1-2):129-46. doi:10.1016/s0166-3542(02)00206-1
- Lipowski D, Popiel M, Perlejewski K, et al. A Cluster of Fatal Tick-borne Encephalitis Virus Infection in Organ Transplant Setting. J Infect Dis. Mar 15 2017;215(6):896-901. doi:10.1093/infdis/jix040
- Wahlberg P, Saikku P, Brummer-Korvenkontio M. Tick-borne viral encephalitis in Finland. The clinical features of Kumlinge disease during 1959–1987. Journal of Internal Medicine. 1989/03/01 1989;225(3):173-177. doi:10.1111/j.1365-2796.1989.tb00059.x
- Kaiser R. Tick-borne encephalitis. Infect Dis Clin North Am. Sep 2008;22(3):561-75, x. doi:10.1016/j.idc.2008.03.013
- Kaiser R. [Tick-borne encephalitis]. Nervenarzt. Jun 2016;87(6):667-80. Frühsommermeningoenzephalitis. doi:10.1007/s00115-016-0134-9
- Bogovic P, Strle F. Tick-borne encephalitis: A review of epidemiology, clinical characteristics, and management. Review. World J Clin Cases. May 16 2015;3(5):430-41. doi:10.12998/wjcc.v3.i5.430
- Haglund M, Günther G. Tick-borne encephalitis–pathogenesis, clinical course and long-term follow-up. Review. Vaccine. Apr 1 2003;21 Suppl 1:S11-8. doi:10.1016/s0264-410x(02)00811-3
- Ruzek D, Avšič Županc T, Borde J, et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Review. Antiviral Res. Apr 2019;164:23-51. doi:10.1016/j.antiviral.2019.01.014
- Dörrbecker B, Dobler G, Spiegel M, Hufert FT. Tick-borne encephalitis virus and the immune response of the mammalian host. Review. Travel Med Infect Dis. Jul 2010;8(4):213-22. doi:10.1016/j.tmaid.2010.05.010
- Simmonds P, Becher P, Bukh J, et al. ICTV Virus Taxonomy Profile: Flaviviridae. J Gen Virol. Jan 2017;98(1):2-3. doi:10.1099/jgv.0.000672
- Pan Y, Cai W, Cheng A, Wang M, Yin Z, Jia R. Flaviviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Front Immunol. 2022;13:829433. doi:10.3389/fimmu.2022.829433
- Labuda M, Jones LD, Williams T, Nuttall PA. Enhancement of tick-borne encephalitis virus transmission by tick salivary gland extracts. Med Vet Entomol. Apr 1993;7(2):193-6. doi:10.1111/j.1365-2915.1993.tb00674.x
- Nuttall PA. Tick saliva and its role in pathogen transmission. Wiener klinische Wochenschrift. 2023/04/01 2019;135(7):165-176. doi:10.1007/s00508-019-1500-y
- Kotál J, Langhansová H, Lieskovská J, et al. Modulation of host immunity by tick saliva. Journal of Proteomics. 2015/10/14/ 2015;128:58-68. doi:10.1016/j.jprot.2015.07.005
- Thangamani S, Hermance ME, Santos RI, et al. Transcriptional Immunoprofiling at the Tick-Virus-Host Interface during Early Stages of Tick-Borne Encephalitis Virus Transmission. Front Cell Infect Microbiol. 2017;7:494. doi:10.3389/fcimb.2017.00494
- Zheng Z, Yang J, Jiang X, et al. Tick-Borne Encephalitis Virus Nonstructural Protein NS5 Induces RANTES Expression Dependent on the RNA-Dependent RNA Polymerase Activity. J Immunol. Jul 1 2018;201(1):53-68. doi:10.4049/jimmunol.1701507
- Hermance ME, Santos RI, Kelly BC, Valbuena G, Thangamani S. Immune Cell Targets of Infection at the Tick-Skin Interface during Powassan Virus Transmission. PLoS One. 2016;11(5):e0155889. doi:10.1371/journal.pone.0155889
- Ellwanger JH, Chies JAB. Host immunogenetics in tick-borne encephalitis virus infection—The CCR5 crossroad. Review. Ticks and Tick-borne Diseases. 2019;10(4):729-741. doi:10.1016/j.ttbdis.2019.03.005
- Shevtsova AS, Motuzova OV, Kuragina VM, et al. Lethal Experimental Tick-Borne Encephalitis Infection: Influence of Two Strains with Similar Virulence on the Immune Response. Front Microbiol. 2016;7:2172. doi:10.3389/fmicb.2016.02172
- Kurhade C, Zegenhagen L, Weber E, et al. Type I Interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by IPS-1. J Neuroinflammation. Jan 27 2016;13:22. doi:10.1186/s12974-016-0487-9
- Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J Virol. Sep 2010;84(17):8470-83. doi:10.1128/jvi.00176-10
- Barkhash AV, Perelygin AA, Babenko VN, et al. Variability in the 2′–5′-Oligoadenylate Synthetase Gene Cluster Is Associated with Human Predisposition to Tick-Borne Encephalitis Virus-Induced Disease. The Journal of Infectious Diseases. 2010;202(12):1813-1818. doi:10.1086/657418
- Kurhade C, Schreier S, Lee YP, et al. Correlation of Severity of Human Tick-Borne Encephalitis Virus Disease and Pathogenicity in Mice. Emerg Infect Dis. Sep 2018;24(9):1709-1712. doi:10.3201/eid2409.171825
- Robertson SJ, Lubick KJ, Freedman BA, Carmody AB, Best SM. Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signaling to inhibit dendritic cell function. J Immunol. Mar 15 2014;192(6):2744-55. doi:10.4049/jimmunol.1302110
- Labuda M, Austyn JM, Zuffova E, et al. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology. May 15 1996;219(2):357-66. doi:10.1006/viro.1996.0261
- Saksida A, Duh D, Lotrič-Furlan S, Strle F, Petrovec M, Avšič-Županc T. The importance of tick-borne encephalitis virus RNA detection for early differential diagnosis of tick-borne encephalitis. Journal of Clinical Virology. 2005/08/01/ 2005;33(4):331-335. doi:10.1016/j.jcv.2004.07.014
- Saksida A, Jakopin N, Jelovšek M, et al. Virus RNA Load in Patients with Tick-Borne Encephalitis, Slovenia. Emerging Infectious Disease journal. 2018;24(7):1315. doi:10.3201/eid2407.180059
- Weber E, Finsterbusch K, Lindquist R, et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. J Virol. Nov 2014;88(21):12202-12. doi:10.1128/jvi.01215-14
- Růzek D, Salát J, Palus M, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. Feb 5 2009;384(1):1-6. doi:10.1016/j.virol.2008.11.023
- Bogovič P, Lotrič-Furlan S, Avšič-Županc T, et al. Comparison of Clinical, Laboratory and Immune Characteristics of the Monophasic and Biphasic Course of Tick-Borne Encephalitis. Microorganisms. Apr 10 2021;9(4)doi:10.3390/microorganisms9040796
- Palus M, Vancova M, Sirmarova J, Elsterova J, Perner J, Ruzek D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology. Jul 2017;507:110-122. doi:10.1016/j.virol.2017.04.012
- Růžek D, Salát J, Singh SK, Kopecký J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One. 2011;6(5):e20472. doi:10.1371/journal.pone.0020472
- Grygorczuk S, Osada J, Toczyłowski K, et al. The lymphocyte populations and their migration into the central nervous system in tick-borne encephalitis. Ticks Tick Borne Dis. Sep 2020;11(5):101467. doi:10.1016/j.ttbdis.2020.101467
- Grygorczuk S, Czupryna P, Pancewicz S, et al. The increased intrathecal expression of the monocyte-attracting chemokines CCL7 and CXCL12 in tick-borne encephalitis. J Neurovirol. Jun 2021;27(3):452-462. doi:10.1007/s13365-021-00975-z
- Toczylowski K, Grygorczuk S, Osada J, et al. Evaluation of cerebrospinal fluid CXCL13 concentrations and lymphocyte subsets in tick-borne encephalitis. Int J Infect Dis. Apr 2020;93:40-47. doi:10.1016/j.ijid.2020.01.023
- Lepej SZ, Misić-Majerus L, Jeren T, et al. Chemokines CXCL10 and CXCL11 in the cerebrospinal fluid of patients with tick-borne encephalitis. Acta Neurol Scand. Feb 2007;115(2):109-14. doi:10.1111/j.1600-0404.2006.00726.x
- Bogovič P, Kastrin A, Lotrič-Furlan S, et al. Comparison of laboratory and immune characteristics of the initial and second phase of tick-borne encephalitis. Emerg Microbes Infect. Dec 2022;11(1):1647-1656. doi:10.1080/22221751.2022.2086070
- Atrasheuskaya AV, Fredeking TM, Ignatyev GM. Changes in immune parameters and their correction in human cases of tick-borne encephalitis. Clin Exp Immunol. Jan 2003;131(1):148-54. doi:10.1046/j.1365-2249.2003.02050.x
- Zidovec-Lepej S, Vilibic-Cavlek T, Ilic M, et al. Quantification of Antiviral Cytokines in Serum, Cerebrospinal Fluid and Urine of Patients with Tick-Borne Encephalitis in Croatia. Vaccines. 2022;10(11):1825.
- Pokorna Formanova P, Palus M, Salat J, et al. Changes in cytokine and chemokine profiles in mouse serum and brain, and in human neural cells, upon tick-borne encephalitis virus infection. J Neuroinflammation. Nov 7 2019;16(1):205. doi:10.1186/s12974-019-1596-z
- Palus M, Vojtíšková J, Salát J, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. Jun 27 2013;10:77. doi:10.1186/1742-2094-10-77
- Blom K, Braun M, Pakalniene J, et al. NK Cell Responses to Human Tick-Borne Encephalitis Virus Infection. J Immunol. Oct 1 2016;197(7):2762-71. doi:10.4049/jimmunol.1600950
- Tomazic J, Ihan A. Flow cytometric analysis of lymphocytes in cerebrospinal fluid in patients with tick-borne encephalitis. Acta Neurol Scand. Jan 1997;95(1):29-33. doi:10.1111/j.1600-0404.1997.tb00064.x
- Grygorczuk S, Świerzbińska R, Kondrusik M, et al. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J Neuroinflammation. Apr 20 2018;15(1):115. doi:10.1186/s12974-018-1138-0
- Michlmayr D, Bardina SV, Rodriguez CA, Pletnev AG, Lim JK. Dual Function of Ccr5 during Langat Virus Encephalitis: Reduction in Neutrophil-Mediated Central Nervous System Inflammation and Increase in T Cell-Mediated Viral Clearance. J Immunol. Jun 1 2016;196(11):4622-31. doi:10.4049/jimmunol.1502452
- Aberle JH, Schwaiger J, Aberle SW, et al. Human CD4+ T Helper Cell Responses after Tick-Borne Encephalitis Vaccination and Infection. PLoS One. 2015;10(10):e0140545. doi:10.1371/journal.pone.0140545
- Schwaiger J, Aberle JH, Stiasny K, et al. Specificities of human CD4+ T cell responses to an inactivated flavivirus vaccine and infection: correlation with structure and epitope prediction. J Virol. Jul 2014;88(14):7828-42. doi:10.1128/jvi.00196-14
- Blom K, Cuapio A, Sandberg JT, et al. Cell-Mediated Immune Responses and Immunopathogenesis of Human Tick-Borne Encephalitis Virus-Infection. Review. Front Immunol. 2018;9:2174. doi:10.3389/fimmu.2018.02174
- Varnaitė R, Blom K, Lampen MH, et al. Magnitude and Functional Profile of the Human CD4(+) T Cell Response throughout Primary Immunization with Tick-Borne Encephalitis Virus Vaccine. J Immunol. Feb 15 2020;204(4):914-922. doi:10.4049/jimmunol.1901115
- Blom K, Braun M, Pakalniene J, et al. Specificity and dynamics of effector and memory CD8 T cell responses in human tick-borne encephalitis virus infection. PLoS Pathog. Jan 2015;11(1):e1004622. doi:10.1371/journal.ppat.1004622
- Lampen MH, Uchtenhagen H, Blom K, et al. Breadth and Dynamics of HLA-A2- and HLA-B7-Restricted CD8(+) T Cell Responses against Nonstructural Viral Proteins in Acute Human Tick-Borne Encephalitis Virus Infection. Immunohorizons. Jul 2 2018;2(6):172-184. doi:10.4049/immunohorizons.1800029
- Gelpi E, Preusser M, Laggner U, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. Aug 2006;12(4):322-7. doi:10.1080/13550280600848746
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol. Jun 2005;64(6):506-12. doi:10.1093/jnen/64.6.506
- Sendi P, Hirzel C, Pfister S, et al. Fatal Outcome of European Tick-borne Encephalitis after Vaccine Failure. Front Neurol. 2017;8:119. doi:10.3389/fneur.2017.00119
- Fujii Y, Hayasaka D, Kitaura K, Takasaki T, Suzuki R, Kurane I. T-cell clones expressing different T-cell receptors accumulate in the brains of dying and surviving mice after peripheral infection with far eastern strain of tick-borne encephalitis virus. Viral Immunol. Aug 2011;24(4):291-302. doi:10.1089/vim.2011.0017
- Varnaitė R. Adaptive Immune Responses to Tick-Borne Encephalitis Virus and SARS-COV-2. Karolinska Institutet (Sweden); 2022.
- Bogovič P, Lusa L, Korva M, et al. Inflammatory Immune Responses in the Pathogenesis of Tick-Borne Encephalitis. J Clin Med. May 22 2019;8(5)doi:10.3390/jcm8050731
- Holzmann H. Diagnosis of tick-borne encephalitis. Review. Vaccine. Apr 1 2003;21 Suppl 1:S36-40. doi:10.1016/s0264-410x(02)00819-8
- Růžek D, Dobler G, Donoso Mantke O. Tick-borne encephalitis: pathogenesis and clinical implications. Travel Med Infect Dis. Jul 2010;8(4):223-32. doi:10.1016/j.tmaid.2010.06.004
- Günther G, Haglund M, Lindquist L, Sköldenberg B, Forsgren M. Intrathecal IgM, IgA and IgG antibody response in tick-borne encephalitis. Long-term follow-up related to clinical course and outcome. Clin Diagn Virol. May 1997;8(1):17-29. doi:10.1016/s0928-0197(97)00273-0
- Stiasny K, Holzmann H, Heinz FX. Characteristics of antibody responses in tick-borne encephalitis vaccination breakthroughs. Vaccine. Nov 23 2009;27(50):7021-6. doi:10.1016/j.vaccine.2009.09.069
- Taba P, Schmutzhard E, Forsberg P, et al. EAN consensus review on prevention, diagnosis and management of tick-borne encephalitis. Eur J Neurol. Oct 2017;24(10):1214-e61. doi:10.1111/ene.13356
- Pierson TC, Diamond MS. Molecular mechanisms of antibody-mediated neutralisation of flavivirus infection. Review. Expert Rev Mol Med. May 12 2008;10:e12. doi:10.1017/s1462399408000665
- Aleshin SE, Timofeev AV, Khoretonenko MV, et al. Combined prime-boost vaccination against tick-borne encephalitis (TBE) using a recombinant vaccinia virus and a bacterial plasmid both expressing TBE virus non-structural NS1 protein. BMC Microbiology. 2005/08/02 2005;5(1):45. doi:10.1186/1471-2180-5-45
- Jacobs SC, Stephenson JR, Wilkinson GWG. Protection elicited by a replication-defective adenovirus vector expressing the tick-borne encephalitis virus non-structural glycoprotein NS1. Journal of General Virology. 1994;75(9):2399-2402. doi:10.1099/0022-1317-75-9-2399
- Khoretonenko MV, Vorovitch MF, Zakharova LG, et al. Vaccinia virus recombinant expressing gene of tick-borne encephalitis virus non-structural NS1 protein elicits protective activity in mice. Immunology Letters. 2003/12/15/ 2003;90(2):161-163. doi:10.1016/j.imlet.2003.09.002
- Kuzmenko YV, Starodubova ES, Shevtsova AS, et al. Intracellular degradation and localization of NS1 of tick-borne encephalitis virus affect its protective properties. Journal of General Virology. 2017;98(1):50-55. doi:10.1099/jgv.0.000700
- Salat J, Mikulasek K, Larralde O, et al. Tick-Borne Encephalitis Virus Vaccines Contain Non-Structural Protein 1 Antigen and may Elicit NS1-Specific Antibody Responses in Vaccinated Individuals. Vaccines (Basel). Feb 12 2020;8(1)doi:10.3390/vaccines8010081
- Timofeev AV, Butenko VM, Stephenson JR. Genetic Vaccination of Mice with Plasmids Encoding the NS1 Non-structural Protein from Tick-borne Encephalitis Virus and Dengue 2 Virus. Virus Genes. 2004/01/01 2004;28(1):85-97. doi:10.1023/B:VIRU.0000012266.04871.ce
- Volpina OM, Volkova TD, Koroev DO, et al. A synthetic peptide based on the NS1 non-structural protein of tick-borne encephalitis virus induces a protective immune response against fatal encephalitis in an experimental animal model. Virus Res. Sep 2005;112(1-2):95-9. doi:10.1016/j.virusres.2005.03.026
- Girl P, Bestehorn-Willmann M, Zange S, Borde JP, Dobler G, von Buttlar H. Tick-Borne Encephalitis Virus Nonstructural Protein 1 IgG Enzyme-Linked Immunosorbent Assay for Differentiating Infection versus Vaccination Antibody Responses. J Clin Microbiol. Mar 25 2020;58(4)doi:10.1128/jcm.01783-19
- Stiasny K, Leitner A, Holzmann H, Heinz FX. Dynamics and Extent of Non-Structural Protein 1-Antibody Responses in Tick-Borne Encephalitis Vaccination Breakthroughs and Unvaccinated Patients. Viruses. May 27 2021;13(6)doi:10.3390/v13061007
- Albinsson B, Rönnberg B, Vene S, Lundkvist Å. Antibody responses to tick-borne encephalitis virus non-structural protein 1 and whole virus antigen–a new tool in the assessment of suspected vaccine failure patients. Infection Ecology & Epidemiology. 2019/01/01 2019;9(1):1696132. doi:10.1080/20008686.2019.1696132
- Ackermann-Gäumann R, Brêchet A, Smetana J, et al. Vaccination against tick-borne encephalitis elicits a detectable NS1 IgG antibody response. J Virol Methods. Dec 2023;322:114831. doi:10.1016/j.jviromet.2023.114831
- Vaccines against tick-borne encephalitis: WHO position paper – Recommendations. Conference Paper. Vaccine. 2011;29(48):8769-8770. doi:10.1016/j.vaccine.2011.07.024
- Holzmann H, Kundi M, Stiasny K, et al. Correlation between ELISA, hemagglutination inhibition, and neutralization tests after vaccination against tick-borne encephalitis. J Med Virol. Jan 1996;48(1):102-7. doi:10.1002/(sici)1096-9071(199601)48:1<102::Aid-jmv16>3.0.Co;2-i
- Agudelo M, Palus M, Keeffe JR, et al. Broad and potent neutralizing human antibodies to tick-borne flaviviruses protect mice from disease. J Exp Med. May 3 2021;218(5)doi:10.1084/jem.20210236
- Füzik T, Formanová P, Růžek D, Yoshii K, Niedrig M, Plevka P. Structure of tick-borne encephalitis virus and its neutralization by a monoclonal antibody. Nat Commun. Jan 30 2018;9(1):436. doi:10.1038/s41467-018-02882-0
- Yang X, Qi J, Peng R, et al. Molecular Basis of a Protective/Neutralizing Monoclonal Antibody Targeting Envelope Proteins of both Tick-Borne Encephalitis Virus and Louping Ill Virus. J Virol. Apr 15 2019;93(8)doi:10.1128/jvi.02132-18
- Baykov IK, Chojnowski G, Pachl P, et al. Structural insights into tick-borne encephalitis virus neutralization and animal protection by a therapeutic antibody. bioRxiv. 2021;
- Heinz FX, Stiasny K. Flaviviruses and their antigenic structure. Review. J Clin Virol. Dec 2012;55(4):289-95. doi:10.1016/j.jcv.2012.08.024
- Tsouchnikas G, Zlatkovic J, Jarmer J, et al. Immunization with Immune Complexes Modulates the Fine Specificity of Antibody Responses to a Flavivirus Antigen. J Virol. Aug 2015;89(15):7970-8. doi:10.1128/jvi.00938-15
- Jarmer J, Zlatkovic J, Tsouchnikas G, et al. Variation of the specificity of the human antibody responses after tick-borne encephalitis virus infection and vaccination. J Virol. Dec 2014;88(23):13845-57. doi:10.1128/jvi.02086-14
- Holzmann H, Vorobyova MS, Ladyzhenskaya IP, et al. Molecular epidemiology of tick-borne encephalitis virus: cross-protection between European and Far Eastern subtypes. Vaccine. 1992/01/01/ 1992;10(5):345-349. doi:10.1016/0264-410X(92)90376-U
- Hayasaka D, Goto A, Yoshii K, Mizutani T, Kariwa H, Takashima I. Evaluation of European tick-borne encephalitis virus vaccine against recent Siberian and far-eastern subtype strains. Vaccine. 2001/09/14/ 2001;19(32):4774-4779. doi:10.1016/S0264-410X(01)00218-3
- Takashima I, Hayasaka D, Goto A, Kariwa H, Mizutani T. Epidemiology of tick-borne encephalitis (TBE) and phylogenetic analysis of TBE viruses in Japan and Far Eastern Russia. Jpn J Infect Dis. 2001/02// 2001;54(1):1-11.
- Rathore APS, St John AL. Cross-Reactive Immunity Among Flaviviruses. Review. Front Immunol. 2020;11:334. doi:10.3389/fimmu.2020.00334
- Mansfield KL, Horton DL, Johnson N, et al. Flavivirus-induced antibody cross-reactivity. J Gen Virol. Dec 2011;92(Pt 12):2821-2829. doi:10.1099/vir.0.031641-0
- Chidumayo NN, Yoshii K, Kariwa H. Evaluation of the European tick-borne encephalitis vaccine against Omsk hemorrhagic fever virus. Microbiol Immunol. Feb 2014;58(2):112-8. doi:10.1111/1348-0421.12122
- Fritz R, Orlinger KK, Hofmeister Y, et al. Quantitative comparison of the cross-protection induced by tick-borne encephalitis virus vaccines based on European and Far Eastern virus subtypes. Vaccine. Feb 1 2012;30(6):1165-9. doi:10.1016/j.vaccine.2011.12.013
- Collins MH, McGowan E, Jadi R, et al. Lack of Durable Cross-Neutralizing Antibodies Against Zika Virus from Dengue Virus Infection. Emerg Infect Dis. May 2017;23(5):773-781. doi:10.3201/eid2305.161630
- Lindblom P, Wilhelmsson P, Fryland L, et al. Factors determining immunological response to vaccination against tick-borne encephalitis virus in older individuals. PLoS One. 2014;9(6):e100860. doi:10.1371/journal.pone.0100860
- Bradt V, Malafa S, von Braun A, et al. Pre-existing yellow fever immunity impairs and modulates the antibody response to tick-borne encephalitis vaccination. NPJ Vaccines. 2019;4:38. doi:10.1038/s41541-019-0133-5
- Rey FA, Stiasny K, Vaney MC, Dellarole M, Heinz FX. The bright and the dark side of human antibody responses to flaviviruses: lessons for vaccine design. EMBO Rep. Feb 2018;19(2):206-224. doi:10.15252/embr.201745302
- Stiasny K, Medits I, Roßbacher L, Heinz FX. Impact of structural dynamics on biological functions of flaviviruses. Review. Febs j. Mar 5 2022;doi:10.1111/febs.16419
- Stiasny K, Kiermayr S, Holzmann H, Heinz FX. Cryptic properties of a cluster of dominant flavivirus cross-reactive antigenic sites. J Virol. Oct 2006;80(19):9557-68. doi:10.1128/jvi.00080-06
- Baldovin T, Mel R, Bertoncello C, et al. Persistence of immunity to tick-borne encephalitis after vaccination and natural infection. J Med Virol. Aug 2012;84(8):1274-8. doi:10.1002/jmv.23313
- Remoli ME, Marchi A, Fortuna C, et al. Anti-tick-borne encephalitis (TBE) virus neutralizing antibodies dynamics in natural infections versus vaccination. Pathog Dis. Mar 2015;73(2):1-3. doi:10.1093/femspd/ftu002
- Kriz B, Hubalek Z, Marek M, Daniel M, Strakova P, Betasova L. Results of the Screening of Tick-Borne Encephalitis Virus Antibodies in Human Sera from Eight Districts Collected Two Decades Apart. Vector Borne Zoonotic Dis. Aug 2015;15(8):489-93. doi:10.1089/vbz.2014.1747