Chapter 9:
TBE in adults
Johann Sellner, Petra Bogovic, Joanna Zajkowska
Key points
- Tick-borne encephalitis (TBE) is a viral infectious disease that involves the nervous system.
- Frequently, there is a febrile illness phase 1-21 days before the onset of neurological symptoms.
- The most common neurological manifestations include meningitis, encephalitis, myelitis, radiculitis, or a combination thereof.
- Long-term sequelae are present in almost every second person with nervous system involvement in this vaccine-preventable disease.
Introduction
Tick-borne encephalitis (TBE) encompasses various diseases caused by infection with the TBE virus (TBEV). TBEV is a positive-strand RNA virus in the genus Flaviviridae, which is primarily transmitted by infected ticks (primarily genus Ixodes) and occasionally by consuming unpasteurized dairy products from infected ruminants.1 Among the several viral subtypes of TBEV, the European subtype (TBEV-Eur) is predominantly found in Europe. Siberian (TBEV-Sib) and Far Eastern (TBEV-FE) are additional prominent subtypes.
An overall increase in TBE cases in the European Union (EU)/European Economic Area (EEA) was observed between 2012 and 2020, according to the European Centre for Disease Control (ECDC).2 In 2021, there was a slight decrease of cases compared to 2020. The drivers of the rising incidence remain unclear.3 For 2021, 22 EU/EEA countries reported 2,949 confirmed cases, with Czechia (n=589), Sweden (n=533), and Germany (n=417) as the front runners. The notification rate was highest in Lithuania (13.1 cases per 100,000 population), followed by Latvia (11.7) and Estonia (6.2). Among the confirmed cases in which information for vaccination was available, 93.2% were not vaccinated against TBE. There is a seasonal pattern for occurrence. In 2021, 90% of confirmed cases occurred between June and November in the EU/EEA, with July being the month with the highest number of reported cases.2
The clinical manifestation of TBE depends on the virulence of the pathogen and the immune status of the host. The majority of the infected people remain asymptomatic or suffer from a self-limiting febrile illness. Some patients develop neurological and neuropsychiatric disturbances caused by meningitis, encephalitis, myelitis, radiculitis, or combinations thereof.4 Cases of nervous system manifestation are more frequently reported among men (male-to-female ratio 1.5:1) and in the age group 45–64 years.2 Mortality of TBE caused by TBEV-Eu is in the range of 0.5-2%. Involvement of the nervous system is associated with long-term sequelae in almost every second survivor.5 Clinical course and long-term outcome vary by TBE virus subtype, although some of the reported differences could be related to access to medical care or testing or methodologic biases.6 Preventive strategies include vaccination and avoiding tick bites; no antiviral medication has been approved.
Risk factors
Ecological variables
TBEV transmission is affected by place, time, and tick population density. However, infection rates in TBE virus–endemic areas are inconsistent, which impedes risk assessments.6 People with outdoor occupations, e.g., farmers, forestry workers, and training in forested areas, are at increased risk for contracting TBE. The risk for TBEV infection for an individual traveler is greatly affected by their itinerary and activities. Among the ECDC cases of 2021, only 1.6% were associated with travel.2 Most infections result from tick bites acquired in forested areas while bicycling, birdwatching, camping, fishing, hiking, or collecting berries, flowers, or mushrooms.6 In contrast, the risk is negligible for people who remain in urban or unforested areas and do not consume unpasteurized dairy products.
Epidemiological data from different European countries demonstrate that the incidence of TBE is higher in older adults than in younger age groups. More than half of the patients are ≥50 years of age.7-9 Both a decline in adaptive and innate immunity and changed lifestyle habits may contribute to this observation.10 This age distribution is also present among TBE cases in vaccinated people.11
Risk factors for severe or protracted course
The most endangered groups for severe clinical manifestation are older adults.12-15 Immunosuppression is another risk factor for unfavorable outcomes. The case fatality rate for TBE is higher in these patient groups.16 A report on a cluster of TBE in organ transplant recipients underscores the association between host immune suppression and fatal outcomes.17 Whether vaccination breakthrough TBE is associated with more severe disease is a matter of investigation.18 A recent study reported that a severe disease course was associated with a low serum TBEV-specific IgG antibody response at the time of onset of the neurologic phase of the disease.19 Another factor that may result in a more severe clinical picture of TBE is the relatively rare occurrence of co-infection with other tick-borne pathogens like Borrelia burgdorferi, Anaplasma phagocytopilum, Rickettsia spp. or Listeria monocytogenes.20,21
Host genetic risk factors
Clinical and epidemiological data indicate that human susceptibility to clinical TBEV infection greatly varies according to age and gender. Mouse models of TBE corroborate that genetic control influences the clinical course of TBE. In this regard, sufficient neutralizing antibody response might be crucial for preventing host fatality. In addition, high expression of various cytokines/chemokines during TBE can mediate immunopathology and be associated with a more severe course of infection and increased fatality.22 Genetic polymorphisms and immune signatures that may predispose to TBEV infection and its severity are covered in the following sections.
The CCR5 plays a crucial role in leukocyte migration and attraction. In human immunodeficiency virus (HIV) infections, the CCR5Δ32 mutation is crucial for invading CD4 cells by HIV particles with a CCR5 tropism.23 In mouse models for flaviviral infections, homozygote CCR5-deficient (-/-) mice died in almost 100% of all infections with West Nile virus (WNV), whereas CCR5 (-/+) heterozygote mice, and homozygote mice with a wildtype CCR5 receptor, had a significantly lower mortality rate.22 These observations from animal studies could be corroborated during a WNV outbreak by identifying the CCR5Δ32 mutation as a strong predictor for a severe clinical disease course in humans. Following the epidemiological results from WNV research, a potential effect of the CCR5Δ32 mutation on TBE was investigated. A clinical study from Lithuania analyzed the incidence of the CCR5Δ32 mutation in different patient populations and found individuals homozygous for CCR5Δ32 only among patients with TBE.24 Moreover, the CCR5Δ32 allele prevalence also increased with the clinical severity of the disease. In another study by this author group, the prevalence of CCR5Δ32 homozygotes was higher in children (2.5%), in adults with severe TBE (1.9%), and in the combined cohort of TBE patients (2.3%) than in controls (0%).25 In a Polish study, the blood expression of CCR5 neither differed between the groups nor did it change in the course of TBE.26 The cerebrospinal fluid (CSF) concentration of the CCR ligand CCL5 was increased in TBE, the highest in the most severe presentation and correlated with pleocytosis. In another Polish study, there were 17.6% CCR5Δ32 heterozygotes and 1.5% homozygotes in the TBE cohort, with no statistically significant difference compared to the controls.27
2′-5′-oligoadenylate synthetases (OAS) are a family of interferon-induced enzymes that play an essential role in mammal antiviral defense. Several polymorphisms in the OAS genes correlated with susceptibility and severe forms of Russian TBE.28,29 The authors of these studies also analyzed OAS polymorphisms in different ethnic populations of the Russian Federation.30 The studies revealed that the frequency of these SNPs correlated with the probability of disease after exposure to TBEV. Very low SNP frequencies were detected in Altaians, Khakasses, Tuvinians, and Shorians, groups with a high exposure risk for TBEV in their native habitats. These findings implicate that TBE risk SNPs may have served as selection factors.
A Czech study evaluated whether innate immunity genes predispose to TBE in humans.31 The analysis showed an association of IFIT1 rs304478 SNP and DDX58 rs3739674 and rs17217280 SNPs and TBE in the Czech population.
The IL-28B polymorphism (rs12979860) is associated with an improved sustained virological response upon treatment with antivirals against Hepatitis C virus (HCV).32 Given the close genetic relationship of flaviviral pathogens like HCV and TBEV, the role of the IL-28B and IL-10 polymorphism was investigated in TBEV infections.33 In a study from the Novosibirsk region of Russia, the IL-28B polymorphism (rs8103142, rs12980275) and the IL-10 polymorphism (rs1800872) were associated with higher risk for severe TBE.
Dendritic cell (DC)-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) is a C-type lectin, expressed by DCs and a subpopulation of macrophages, involved in the detection of pathogen-associated molecular patterns (PAMPs), cell migration, and interaction with T lymphocytes, potentially contributing to an early response to TBEV at the site of tick feeding and initiation of a specific immune response.34 Findings in the context of dengue virus and HCV infections pointed to an increased risk of dengue hemorrhagic fever and advanced hepatic injury in hepatitis C when there is an underlying SNP (rs4804803) located in the promotor region of the CD209 gene.30 DCs in the skin and gut may play an important role as antigen-presenting cells and virus spread early in TBEV infection.35 A study from Russia of presumably TBEV-Sib cases showed a correlation between the presence of 2 SNPs (rs4804803, rs2287886) in the promotor region of the CD209 gene and the severity of the TBE disease course.30
MMP-9 directly degrades extracellular matrix proteins and activates cytokines and chemokines to regulate tissue remodeling. In a study of Russian TBE cases, the frequency of the rs17576 G allele of MMP-9 was significantly higher in TBE cases with severe CNS diseases.36
Taken together, several studies disclosed a potential role for various gene polymorphisms in the susceptibility and severity of TBE. These findings need to be corroborated in independent cohorts with appropriate controls, using uniform criteria for disease severity and characterization of the virus strain, as there are also trials that could not confirm these observations.37
Clinical course
Definitions of the clinical presentations and time frames
Infection with TBEV may be symptomatic or asymptomatic. A symptomatic infection may manifest as a febrile illness without nervous system involvement or as TBE (Figure 1).38
Figure 1: Timeline of clinical manifestations after TBEV infection

Asymptomatic infection with TBEV is defined as TBEV IgG antibody seroconversion in an asymptomatic person.
Febrile illness resulting from infection with TBEV is defined by the presence of fever and constitutional symptoms, the absence of signs/symptoms of CNS involvement at the time of actual illness, and the presence of TBEV RNA in serum and/or later seroconversion to TBEV. According to the later appearance (or absence) of neurologic involvement, the febrile illness is further sub-classified as either the initial phase of TBE (defined as a febrile illness that, after a clinical improvement, is followed by neurologic involvement occurring within at least a 1-month follow-up period and fulfilling criteria for TBE) or as febrile illness resulting from infection with TBEV in a narrow sense (abortive form of TBE, febrile headache, summer flu, fever form) when no signs/symptoms of CNS involvement are present at the time of actual illness or within at least a 1-month follow-up period.38
TBE is defined as the presence of clinical signs or symptoms of nervous system involvement (i.e. meningitis, encephalitis, myelitis, radiculitis, or a combination), with increased CSF leukocyte counts (>5 × 106 cells/L), and demonstration of a recent infection with TBEV indicated by serum specific IgM and IgG antibodies or IgG seroconversion in paired serum samples.13,39This definition partly contradicts the ECDC case definition for TBE, which does not explicitly require CSF pleocytosis to diagnose TBE;40 however ECDC definitions are intended for epidemiological monitoring and are not necessarily optimal for clinical use. The approximate time course of TBE is shown in Figure 2.41
Figure 2: Scheme of clinical events and antibody evolution in TBEV infection

Pathogenesis – clinical highlights
After the bite of an infected tick, TBEV replication occurs locally in the subcutaneous tissue. DCs of the skin (Langerhans cells) play an essential role since they bind with antigens and subsequently induce an immune response by producing proinflammatory cytokines. Langerhans cells are the most relevant cell group for local viral replication, transporting the virus to the regional lymph nodes where further replication occurs. After release into the bloodstream from the lymph nodes, TBEV disseminates to other organs, particularly the reticulo-endothelial system (mainly bone marrow, spleen, and liver), where the virus continues to multiply and maintain viremia for several days. Probably during the second viremic phase (which clinically matches with febrile illness without CNS involvement), the virus reaches the brain.42,43 The precise mechanism of viral passage through the blood-brain barrier is unclear but depends on the presence of viremia. There are four candidate routes:
- direct axonal retrograde transport from infected peripheral nerves;
- infection of highly susceptible olfactory neurons;
- virus entry into vascular endothelial cells of brain capillaries, transcytosis, and release of virus into the brain parenchyma; and
- diffusion of virus between capillary endothelial cells.
There is also a so-called “Trojan horse” mechanism, which assumes that the virus is transported by infected immune cells to the CNS.42,44,45 The primary targets of TBEV infection in CNS are neurons. Rarely, oligodendrocytes are infected.42
The pathogenesis of asymptomatic infections in humans is poorly defined. It seems logical that, on the one hand, the virus enters the body similarly to symptomatic infections and, on the other hand, does not enter the CNS. Still, it is not clear whether the development of the disease is deterred or interrupted after multiplication in the lymph nodes before or following penetration into the blood.
The characteristics of the TBEV subtype, the quantity of virus copies, and the host immune response influence the pathogenesis. The immune response is necessary not only for controlling TBEV infection but is also thought essential for the resulting clinical manifestations, but knowledge of such responses is incomplete.41,46 Immune responses during TBEV infection are described in a separate chapter.
Presentations of tick-borne virus infection
1. Asymptomatic infections
Seroepidemiological studies suggest that most TBEV infections (70%‒98%) are asymptomatic; however, the exact proportion of such cases is unknown because probably part of those with mild clinical presentation may remain below the diagnostic threshold. 47-49
2. Symptomatic infections
The time interval from a tick bite to the beginning of the illness is usually 7–14 days, but it may be as short as two days and as long as four weeks. With the alimentary route of infection, there is usually a shorter incubation period of 3 to 4 days; however, the reports are not unanimous.50-55
2.1. Febrile illness due to TBEV infection (abortive form of TBE, febrile headache, summer flu, fever form)
Information on febrile illness due to TBEV infection also called the abortive form of TBE, febrile headache, summer flu, or fever form, is limited. Clinically and serologically, the abortive form of TBE has been postulated to match the initial phase of TBE, except that subsequent CNS involvement does not occur. Because clinical symptoms and signs of the illness are non-specific, and because, in parallel to the initial phase of TBE, serum antibodies to TBEV are not yet expected to have developed, the only option for diagnosis at the time of actual illness is demonstrating the presence of TBEV RNA in the blood. However, this approach is not routine and might have a low diagnostic yield owing to several other known or unknown causes of fever, even in a highly endemic region for TBE. Therefore, the possibility that a febrile illness results from TBEV infection is usually tested for and established only after signs or symptoms of CNS involvement appear, which does not happen in the case of the fever form. In that case (and if PCR detection of viral RNA in blood is unavailable), further clinical and microbiologic (serologic) follow-up after improvement is needed to establish the diagnosis.
Data on the frequency of this clinical manifestation of the disease caused by European TBEV subtype are conflicting. TBEV infection manifesting as febrile illness without later CNS involvement is considered frequent,55-57 although not in all reports,52,58-60 but the scientific basis for such a conclusion is unclear. According to some reports, it represents more than half of all clinically manifested TBEV infections.55,56 However, this is not confirmed by the results of prospective clinical trials on the etiology of acute febrile illness after a tick bite. In the study by Lotric-Furlan and co-workers, among 56 patients diagnosed with TBEV infection by the presence of TBEV RNA in blood by PCR during febrile illness that developed after a tick bite, in 55 (98.2%) CNS involvement with pleocytosis later appeared.61,62 In contrast, only one (1.8%) had an isolated febrile illness without later CNS involvement. A more recent, similarly designed study from Slovenia revealed that illness progressed to TBE in 52/62 (84%) adult patients within 18 days after defervescence.38 In the Russian literature, this clinical manifestation is named “fever form” and is reported to represent up to 50% of all clinical presentations of TBEV infections.63
The current view is that febrile illness caused by TBEV infection most frequently presents as a moderate fever, headache, fatigue, and other non-specific symptoms and clinically corresponds to the initial phase of the TBE. The fever usually resolves in a few days, and the disease does not have long-term consequences.38,64,65 The outcome of symptomatic TBEV infection without CNS involvement is believed to be favorable; however, very little reliable information on the outcome has been published.38
2.2. Tick-borne encephalitis
In 56–87% of symptomatic patients infected with the European subtype of TBEV, CNS inflammation is preceded by a febrile illness, resulting in a biphasic course of the disease.4,12,19,52,53,60,66-69 The initial illness (first phase of TBE), which corresponds to viremia, presents with fever, fatigue, malaise, headache, and muscle and joint pain that occurs without CNS inflammation. It usually lasts less than one week, followed by improvement lasting several days.38,53,70 The hallmark of the second phase of TBE is CNS involvement: in approximately 50% of adult patients, it presents as meningitis, in about 40% as meningoencephalitis, and around 10% as meningoencephalomyelitis.49 The frequency of different neurological presentations has been somewhat variable.9,53,60,68,71
Some patients with TBE have no (obvious) initial phase of the disease and present directly with central nervous system involvement. Data on the monophasic course of the disease are incomplete. Some studies showed that patients with monophasic presentation of TBE have a more severe clinical course of the disease than those with biphasic course.12,52,53 In addition, some reports on patients with severe TBE who needed intensive care management show an unusually high proportion of those with monophasic course (15/31, 48.4% and 21/33, 63.6%, respectively).72,73 A direct comparison of the clinical presentation and laboratory findings in 705 adult TBE patients, of whom 283 had monophasic and 422 had biphasic course, revealed that patients with the monophasic course were significantly older, more often vaccinated against TBE (7.4% vs. 0.9%), more often had comorbidities (52% vs. 37%), and were more often treated in the intensive care unit (12.4% vs. 5.2%). However, the long-term outcome 2–7 years after TBE was comparable.74
Case fatality rate in TBE caused by the European subtype of TBEV is 0.5–2% and generally increases with age.49,70
TBE caused by Far-Eastern TBEV subtype has been characterized with more severe disease and a case fatality rate of up to 40%, while in TBE caused by Siberian virus subtype the reported case fatality rate is 2–3%, and cases of chronic and progressive forms have been described.48,70,75,76
The initial phase of tick-borne encephalitis
Information on the initial phase of TBE is limited. Characterization of 98 adult patients who had TBEV RNA in their blood but no CNS involvement at the time of evaluation revealed that incubation (time from tick bite to onset of the illness) was six days, median duration of illness was seven days, and that 37 (38%) patients were hospitalized for a median three days. The most frequent findings were malaise or fatigue (98%), fever (97%), headache (86%), and myalgia (54%), followed by arthralgia (43%), gastrointestinal symptoms (46%; abdominal pain 2%, nausea/vomiting 38%, loose stools 16%), respiratory symptoms (18%; sore throat 11%, cough 10%) and chills (19%). Typical laboratory findings were leukopenia (88%), thrombocytopenia (59%), and abnormal liver function test results (63%). At the time of positive PCR findings, 0/98 patients had serum IgG TBEV and seven serum IgM TBEV; all patients later seroconverted. Viral RNA load was higher in hospitalized patients with more severe illness than in those who did not need hospitalization but did not differ substantially according to age, sex, duration of illness before testing, or total duration of the actual febrile illness, or for patients with undetectable viral IgM in serum samples when compared with patients in whom antibodies were detectable. Illness progressed to TBE in 84% within 18 days after defervescence.38 Clinical and laboratory findings in patients with TBEV febrile illness did not distinguish between patients in whom TBE later develops and those in whom it does not.
Clinical spectrum of neurological manifestations
Meningitis is characterized by fever, headache, nausea, vomiting, and meningeal signs. These symptoms and signs are present in most patients but not all. In a study encompassing 448 adult patients with TBE from Slovenia, almost all reported headache and had fever, more than 50% suffered from nausea and/or vomiting, and 70% had clearly expressed meningeal signs.68
Encephalitis may manifest by a variety of neurological symptoms and signs, most often with tremor (especially of the fingers of the upper extremities and tongue), sometimes with nystagmus, speech disorder, ataxia, and movement disorders, occasionally with seizures, and rarely with brain stem symptoms and/or cranial nerve abnormalities. Impaired consciousness, ranging from mild to severe, concentration disturbances, and cognitive function disturbances are rather frequent; amnesia, behavioral changes, psychosis, and delirium may also occur.
Myelitis manifests with flaccid paralyses that are occasionally preceded by severe pain in the affected muscle groups. The involvement is usually asymmetrical. Most often, the extremities are affected, more frequently the upper than the lower limbs, and more often the proximal segments of the extremities than the distal ones. Patients with pareses of respiratory muscles usually require artificial ventilatory support.13,39,52,53,60
Radiculitis is a rare manifestation of TBE.77 In patients with TBE who have radiculitis it is reasonable to look for concomitant Borrelia infection.
Other manifestations in the acute phase of tick-borne encephalitis
Involvement of cranial nerves. Involvement of cranial nerves is rare (usually in less than 5% of patients), mainly asymmetrical, and usually has a favorable outcome. Ocular, facial, and pharyngeal muscles are most often affected, but hearing and vestibular defects are also encountered.4,9,52,53,60 In a series of 1218 adult patients diagnosed with TBE at a single center, 11 (0.9%) developed peripheral facial palsy (two bilateral, nine unilateral); and occurs late in the course of acute illness(10-20 days after the onset of the neurological phase and more frequent in patients with an encephalitic than a meningitis manifestation of TBE. However, 3 out of 11 patients had associated borrelial infection 10.1111/j.1469-0691.2011.03719.x. The latter finding suggests that in patients who develop peripheral facial palsy in the course of TBE, and who had been exposed to ticks in the region where both TBE and Lyme borreliosis are endemic, coexistent infection with Lyme borreliosis has to be taken into account.78
Autonomic nervous system disorders. Occasionally, autonomic nervous system disorders occur in patients with TBE. These include cardiac and enteric nervous system disturbances.79,80
Encephalitis with normal CSF cell count
There are a few reports on a serologically confirmed TBEV infection in TBE but without CSF pleocytosis.81,82 This observation disagrees with the large series of serologically proven TBE patients in which CSF pleocytosis was found in all cases.13,39,53 However, the latter findings might result from a selection bias because CSF pleocytosis was one of the essential inclusion criteria for the diagnosis of TBE.
3. Chronic progressive tick-borne encephalitis
There is no agreement on the existence of chronic TBE. Cases of a chronic progressive form of TBE were reported from Siberia and the Russian Far East, caused by the Siberian TBEV subtype. Both mutations in the TBEV NS1 gene and an inappropriate T-cell immune response are implicated in chronic progressive disease.70 According to information from Western Siberia, 1.7% of patients with acute TBE develop a chronic progressive form of the disease.83 Clinical presentations include Kozshevnikov’s epilepsy, lateral sclerosis, progressive neuritis, progressive muscle atrophy, and a Parkinson-like disease. A broad spectrum of incubation periods, time to the onset of individual neurological signs/symptoms, and survival after the onset of the disease have been reported.84,85 Progressive TBE is probably not present or uncommon in diseases caused by European TBEV subtype. In the study carried out in Lithuania, where only European TBEV subtype has been recorded, the progressive course was noted in two out of 133 consecutive patients with acute TBE.53,86
TBE in particular situations (in immunocompromised persons, during pregnancy, in persons vaccinated against the disease) is presented in another chapter(s).
Laboratory findings
CSF pleocytosis
CSF pleocytosis is a dominant laboratory finding in patients with TBE. In 2 large studies, encompassing 731 and 717 adult patients with TBE, respectively, the median leukocyte values were 60 × 106/L, with a maximal count of 1200 ×106/L.13,87 Some studies indicate that CSF leukocyte count is lower in persons with TBE who are older than 60 years than in younger adults.68 Lymphocytic predominance in CSF is typical for TBE; however, granulocytes may prevail during the first few days (Figures 3 and 4). Most patients have mild to moderately elevated protein and albumin concentrations in CSF and elevated albumin and IgG indexes, indicating disruption of the blood-brain barrier.13,68,70,88
Figure 3: Evaluating pleocytosis in TBE (early)
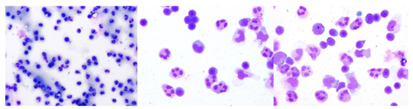
Lymphocyte populations in CSF during acute TBE: The pleocytosis is dominated by mononuclear cells (lymphocytes and monocytes). Polymorphonuclear cells may predominate at a very early stage. (1 x 100; 1 x 400; 1 x 400.)
Figure 4: Evaluating pleocytosis in TBE (later)
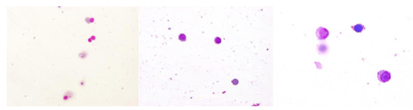
During recovery, after acute phase, control LP. x 100, single lymphocytes, some monocytes, lack of granulocytes x 200 x 400.
Peripheral blood
Laboratory abnormalities in the blood are more pronounced in the initial phase of TBE (and in the abortive form of the disease) than in the meningoencephalitic phase. In the first phase of TBE, the number of leucocytes in the peripheral blood is frequently reduced, while in the second phase, it is normal or slightly elevated. Furthermore, the initial phase is characterized by thrombocytopenia and elevated liver enzymes, while the second phase is not; moreover, inflammatory markers are usually within normal limits in the first phase of the disease but may be slightly elevated in some patients in the second phase.38,39,52,70,89,90 The differences are best shown by comparing the results in patients assessed for laboratory abnormalities in the first and second phases of the disease. An example of such an approach is an analysis of 88 patients with biphasic course of TBE, in whom TBEV RNA in blood was established during the initial phase of illness and who later developed CNS inflammation and seroconversion. Comparison of laboratory findings in the initial and the second (meningoencephalitic) phase of TBE in this study revealed significant differences in peripheral blood leukocyte counts (including neutrophil, lymphocyte, and monocyte counts) and platelet counts, as well as serum concentrations of C-reactive protein, aspartate aminotransferase, and gamma-glutamyl transferase but not for alanine aminotransferase (Table 1).89 A recent study exposed that in addition to previously known leukopenia, thrombocytopenia, and increased liver enzymes, the initial phase of TBE is relatively often associated also with elevated muscle enzyme activities: 33% of patients had elevated serum creatine kinase, 26% myoglobin and 22% troponin activity; at least one of the muscle enzymes was elevated in 42% of patients. Leukopenia, thrombocytopenia, elevated liver enzymes, and elevations of creatine kinase and myoglobin were present in the initial phase but resolved later, while mild troponin abnormalities were also found in the second phase of TBE.91
Table 1: Comparison of laboratory findings in patients with the initial and the second(meningoencephalitic) phase of tick-borne encephalitis.89
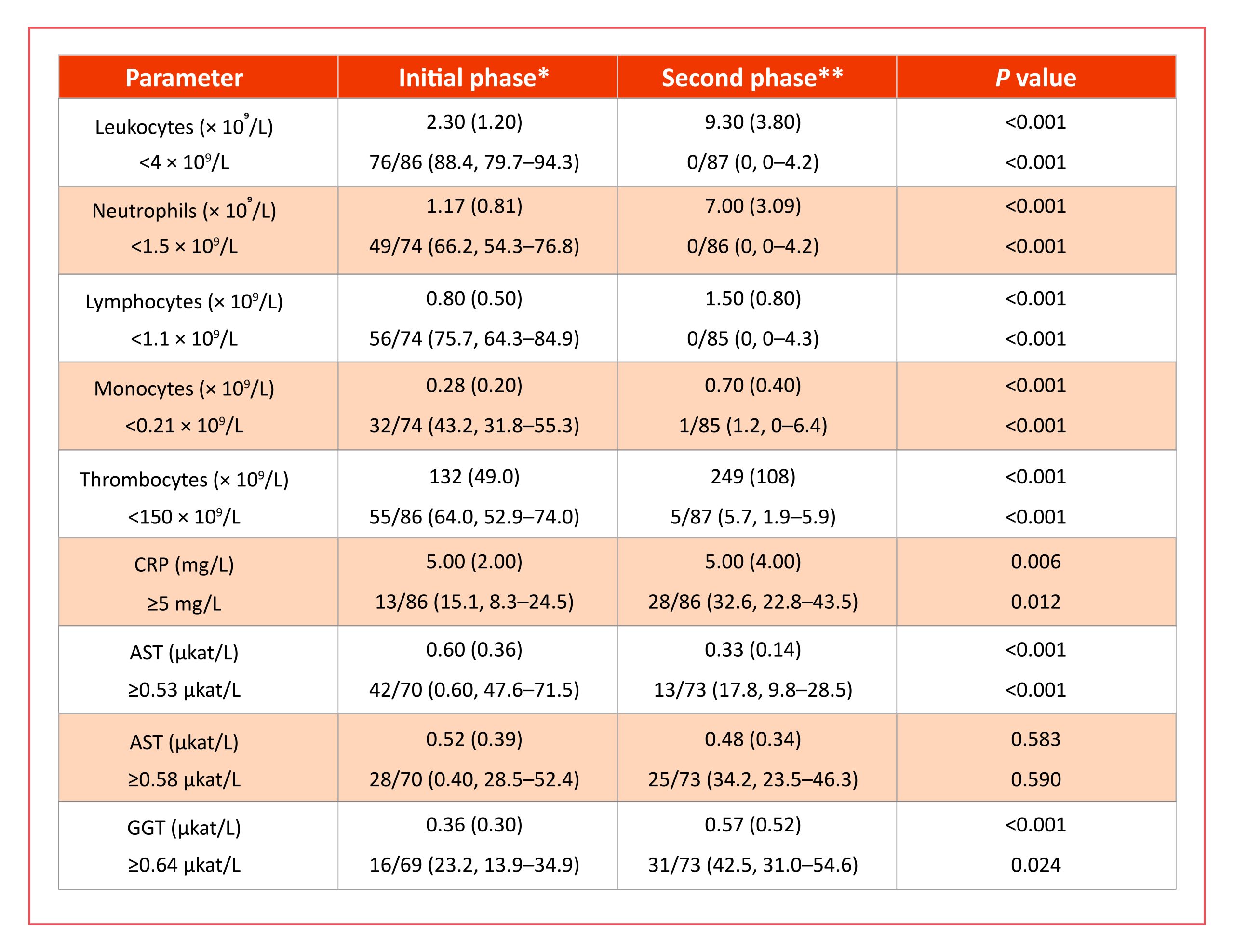
Data are given as median (interquartile range) or proportion (%, 95% confidence interval). CRP, C-reactive protein; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma glutamyl transferase. * Median duration of illness 5 days, range 1–10 days; ** Median duration of meningoencephalitic phase of illness 2 days, range 1–10. Median symptom free interval 8 days.
Neuroimaging
Neuroimaging enables rapid, non-invasive visualization of the central and peripheral nervous system. In clinical practice, neuroimaging is indispensable to corroborate clinical suspicion of nervous system inflammation, rule out mimics, provide hints for the causative pathogen, and assess for complications. Magnetic resonance imaging (MRI), with its excellent soft tissue contrast, is superior to computed tomography (CT). CT is used for exploratory examination of the brain on admission, in case of rapid clinical deterioration, and before lumbar puncture.
The nervous system manifestations of TBEV infection include meningitis, encephalitis, myelitis, and radiculitis.4 Most changes in neuroimaging of viral encephalitis are unspecific. They can be observed with several other pathogens and neurological disorders.92 Some radiological features are shared across infectious, immune-mediated, and non-inflammatory causes of nervous system disorders.93 Moreover, radiological signs may be absent despite clinical signs and symptoms of meningeal, parenchymal, spinal cord, or peripheral nervous system dysfunction. Studies on the correlation of clinical severity with imaging findings are not available in TBE.
Meningitis
Clinical features of meningitis encompass the classic triad of fever, nuchal rigidity, and nausea/vomiting. Meningitis primarily involves the leptomeninges, which consist of the inner arachnoid and the pial meningeal layers. Unenhanced CT can display mild dilatation of the ventricles with effaced subarachnoid spaces, suggesting diffuse cerebral swelling.94 MRI is more sensitive for detecting radiological features of meningitis than CT.95 T1-weighted MR imaging may show obliteration of the basilar cisterns. Fluid-attenuated inversion recovery (FLAIR) sequences may demonstrate hyperintensity in the subarachnoid space, even when T1-weighted images appear normal. Postcontrast T1-weighted images may show linear continuous sulcal or cisternal enhancement, with predilection at the basal meninges and cerebellar folia.96 Enhanced and thickened cranial nerves may also be observed.97
Encephalitis
Encephalitis is defined as inflammation of the brain parenchyma associated with neurologic dysfunction. MRI is essential in diagnosing encephalitis, evaluating the disease course and complications, and prognosis.98 Encephalitic lesions of TBE are present as areas of increased signal intensity on T2/FLAIR-imaging (Figures 5, 6, and 7), which may also enhance upon administration of contrast agents.99,100 In TBE, the enhancement is mainly restricted to the lesion margins.96
Figure 5: MRI visualization of TBE-related abnormalities
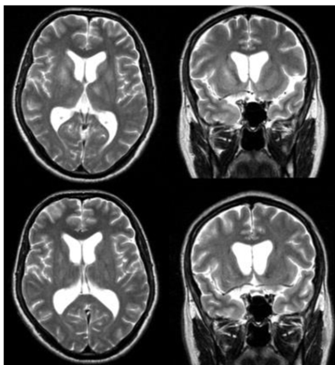
Axial (A) and coronal (B) T2-weighted MRI images show high signal intensity in the basal ganglia and thalami. The second scans (C, D) obtained several months later, show partial resolution of the lesions. Patient with chorea presentation.
Figure 6: Further visualization of TBE-related abnormalities
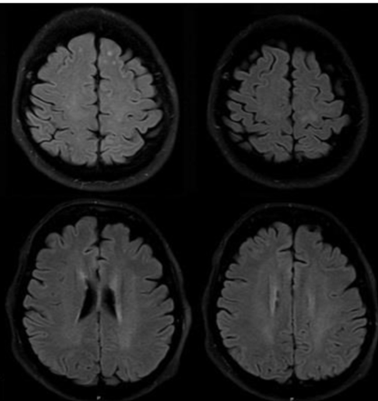
Axial FLAIR images. There is abnormal signal intensity in the left frontal (A) and left parietal lobe (B) and confluent, poorly visible abnormal bilateral hyperintensity in the periventricular white matter (C) and in the centrum semiovale (D). Parkinsonism as residual sequelae.
Figure 7: Additional visualization of TBE-related abnormalities
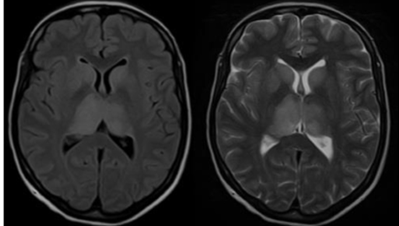
Axial fluid-attenuated inversion recovery (FLAIR) image (A) and T2-weighted MR image (B) show bilateral hyperintensity of the caudate nuclei, putamina and thalamus. The right side is slightly more involved than the left side. Patient with immunosuppression.
The sensitivity of MRI to detect brain lesions despite clinical symptoms of encephalitis due to TBEV infection is low. In a Swiss study of patients with encephalitis or meningoencephalitis by TBE and MR imaging performed after a median of 10 days, 27% had lesions on FLAIR and 6% diffusion restrictions.100 Leptomeningeal enhancement was detected in 44% and brain hemorrhage in 5%. Even with repeated scans, the yield for detecting parenchymal damage in patients with an encephalitic syndrome was 46%, according to an Austrian study.18 The time point of imaging could play a significant role in this regard. Brain lesions were detected in two patients on day 21 from hospital admission in the latter study, whereas these were not present on the scans on days 5 and 8, respectively. Contrast enhancement is found only in the minority of patients.18
The predilection sites of brain lesions in TBE on FLAIR were the thalamus (50%) and the pontine area (29%) in the Swiss study.100 Thalamic lesions can be uni- or bilateral. Lesions were less frequent in the limbic regions (amygdala and hippocampus, each 21%), the mesencephalon, and the cerebellum (each 21%). In the Austrian study, the predilection sites were the periaqueductal grey (17%), the thalamus, and the brainstem (each 12%).18 Among the patients in whom a brain lesion was detected, the median number of lesions was 2. In a pilot study of patients with an encephalitic TBE course, glucose hypometabolism was present in 7 out of 10 TBE patients at sites prone to lesion development.101 Glucose hypometabolism reflects neuronal dysfunction and did not correlate with MRI brain lesions due to TBEV. In line, MR spectroscopy of TBE lesions during the acute phase of the disease shows changes indicative of necrosis. The presence of brain lesions on MRI and lesion expansion may determine prognosis.18,100 The persistence of lesions over time has not been studied systematically so far. There is anecdotal evidence of a complete resolution of cerebral, brainstem, and spinal cord lesions within six months.102 A Polish study of patients with encephalitic lesions during acute TBE studied structural brain changes 12 months later.103 On follow-up, there was marked brain atrophy with a widening of the anterior horns and lateral ventricles, indicating grey and white matter loss.
Myelitis and radiculitis
Myelitis and radiculitis with TBEV infection can occur isolated or in combination. Spinal cord and nerve root MRI findings were studied only in smaller patient series and case reports. TBEV has a propensity for the anterior horn cells of the grey matter in the spinal cord.96 These lesions are commonly longitudinally extensive, defined as an expansion over three or more vertebral segments, and can expand to the brainstem.104 Both uni- and bilateral lesions of the grey matter have been reported and are associated with a Polio-like syndrome characterized by acute flaccid paresis.105,106 There can be a swelling of the grey matter and lesional and leptomeningeal contrast enhancement.104 Spinal cord lesions often enhance markedly.96 Rarely, the posterior horns may also be involved.96 In radiculitis, the roots of the spinal nerves may be thickened and display contrast enhancement.77,107
Electroencephalography
For viral encephalitis, electroencephalography (EEG) is a valuable adjunct to clinical neurological examination. It can detect subtle or subclinical disturbances of cerebral function and enables the detection and monitoring of seizure activity over time.108 In most cases, the EEG findings are non-specific and denote global compromise of the brain function but may also provide information about prognosis and therapeutic response. Abnormal EEG findings were reported in 77% of patients with TBE.69 In most cases, an initially abnormal EEG normalizes within a few weeks. However, a small study of children with TBE reported a higher likelihood of impaired attention and psychomotor speed and that the EEGs were significantly slower on follow-up than control EEGs.109
Epileptic seizures can occur as the initial manifestation or during TBE.71,110 Continuous EEG monitoring for at least 48 hours is recommended in patients with persistent unconsciousness to evaluate intermittent non-convulsive seizures or even persistent non-convulsive status epilepticus.39 The 10-year risk of epilepsy after TBE is 1.7% (95% CI 0.7-2.7).111
Prognosis and long-term sequelae
The analysis of the standardized mortality ratio (SMR) in Sweden from 2004–2017 revealed a mortality rate for TBE infection to be ≈4-fold higher than that of the matched control population.112 The SMR was 3.96 (95% CI 2.55–5.90). The case fatality rate (CFR) was 0.75% in this study and in the range of previously reported rates of 0.5% in Europe.113 No cases in patients <40 years of age were fatal. CFR for diseases caused by the two non-European TBEV subtypes is generally higher, but the data are very limited. In lethal cases, death occurs within 5–10 days after the onset of neurological symptoms in the context of diffuse brain edema or bulbar involvement.
TBE is associated with individual and societal disease burden. The need for hospital care is increased, with protracted in-hospital stays and admission to the intensive care unit during acute TBE.39 Moreover, the study of the Swedish National Health Data Register for TBE cases diagnosed during 1998-2014 revealed that patients with TBE were hospitalized for more days during the first year after disease onset (11.5 vs. 1.1 days) and had more specialist outpatient visits (3.6 vs. 1.2 visits).114 They also had more sick leave days (66 vs. 10.7 days) than a reference cohort without TBE, indicating significant productivity losses.
The high proportion of patients with persistent post-TBE symptoms is another strong argument for preventive strategies. Sequelae can be categorized as neurological (e.g., paresis, limb paresis, aphasia, ataxia, sensory impairment, epilepsy, tremor, hearing disorder), neuropsychiatric symptoms (e.g., concentration and memory deficits), and general/unspecific (fatigue, headache, general weakness, poor sleep quality, sweating disturbances), as summarized (Table 2). Previous prospective studies disclosed that neurological and neuropsychological sequelae persist in 40–46% of the patients one year after the acute phase of the disease.53,60 A study from Slovenia reported that the rate of persistent symptoms was higher at six months than at 12 months, which points to some improvement and regenerative capacity within the first year after TBE.115 Recent studies corroborate the rate of incomplete recovery beyond 12 months. A study from Southern Germany performed telephone interviews after 18 months from TBEV infection; the period was 2018 to 2020.14 Full recovery was reported by 67.3% (children: 94.9%, adults: 63.8%). Sequelae included fatigue (17.0%), weakness (13.4%), concentration deficit (13.0%), and impaired balance (12.0%). The recovery rate was 64% lower after severe TBE (compared to mild; HR: 0.36, 95%CI 0.25-0.52) and 22% lower with comorbidities (HR: 0.78, 95%CI 0.62-0.99). Substantial healthcare use was reported (90.1% hospitalization, 39.8% rehabilitation). A study from Lithuania evaluated long-term neurological and neurocognitive sequelae after TBE in adults.116 This prospective study from 2018-2019 revealed that 25.5% of the patients had moderate or major impairment (Glasgow Outcome Scale, GOS) and various levels of disability in 34.7% (Rankin-Scale, RS) at discharge. Up to 18 months from the onset of TBE, over 20% remained with slight to moderate disability (modified RS, mRS). GOS, RS, and mRS scores correlated with disease severity.
Table 2: Overview of main sequelae and affected domains in TBE, adapted from5

There is also evidence for the development of post-encephalitic syndrome (PES). Some authors define PES as the presence of ≥ 2 subjective symptoms that developed or worsened since the onset of TBE and had no other known medical explanation and/or ≥ 1 objective neurological sign.115
The reporting of sequelae is affected by a lack of standardized reporting. Consensus criteria for classifying sequelae of TBE and its severity are eagerly awaited. Such a reporting system should include neurological and neuropsychological examinations for the evaluation of cerebral symptoms as well as a scoring system for spinal cord and peripheral nervous system disturbances. A harmonized classification system would also be helpful for a better understanding and monitoring of PES.
Treatment
No specific antiviral therapy is currently available and approved for TBEV infections. Some antiviral agents, specific immunoglobulins, and other potentially protective substances are under investigation for their anti-TBEV efficacy117; however, a detailed review of these ‘pipeline’ agents is beyond the scope of this chapter.
Treatment is supportive and symptomatic. Fever is associated with increased metabolic consumption and dehydratation. Antipyretics, or other physical measures like cooling blankets, or infusion of cooled fluids, should be employed to reduce body temperature. TBE can be accompanied by hypovolemia due to a decreased intake and a secondary loss of fluids. Hyponatremia is a common condition in patients with TBE, including the syndrome of inappropriate antidiuretic hormone secretion (SIADH), cerebral saltwasting syndrome, and reduced sodium supplementation.118 Mental and behavioral disturbances, delirium, and psychotic signs and symptoms may justify treatment with neuroleptics. In line with other types of brain injury, primary prophylaxis of seizures is currently not recommended, and treatment of clinical seizures is based on general guidelines for the management of seizures/status epilepticus. Pain and arousal cause intracranial pressure peaks by increasing the cerebral blood flow; therefore, sedatives and careful clinical monitoring are key factors in the prevention of intracranial hypertension and its complications.
Encephalitis often requires ICU admission to ensure oxygenation, airway protection, circulatory support, and prevention and treatment of secondary complications that may impact outcomes. These include cerebral edema, seizures/status epilepticus, and systemic complications, such as fever, aspiration pneumonia, and respiratory failure requiring mechanical ventilation is also needed in patients with severe respiratory muscle paresis, in some cases lifelong.119 Early recognition of complications and admission to the ICU is crucial for improving prognosis.
Most survivors do not recover fully and often require extended posthospitalization rehabilitation and care to regain their functional abilities.5 A comprehensive assessment of neurological, cognitive, and psychiatric functions after hospital discharge is mandatory. Moreover, referral to rehabilitation services and psychiatric support, as with other neurological disorders, is indicated to improve the quality of life of both the patient and their caregivers.
Contact
Johann Sellner
johann.sellner@mistelbach.lknoe.at
Petra Bogovic
petra.bogovic@kclj.si
Joanna Zajkowska
joanna.zajkowska@umb.edu.pl
Affiliation
Johann Sellner, Petra Bogovic, Joanna Zajkowska
Citation
Sellner J, Bogovic P, Zajkowska J. TBE in adults. Chapter 9. In: Dobler G, Erber W, Bröker M, Chitimia-Dobler L, Schmitt HJ, eds. The TBE Book. 7th ed. Singapore: Global Health Press; 2024. doi:10.33442/26613980_9-7
References
- Chiffi G, Grandgirard D, Leib SL, Chrdle A, Ruzek D. Tick-borne encephalitis: A comprehensive review of the epidemiology, virology, and clinical picture. Rev Med Virol. Sep 2023;33(5):e2470. doi:10.1002/rmv.2470
- ECDC. Tick-borne encephalitis. ECDC Annual epidemiological report. European Centre for Disease Prevention and Control; 2024.
- Saegerman C, Humblet MF, Leandri M, et al. First Expert Elicitation of Knowledge on Possible Drivers of Observed Increasing Human Cases of Tick-Borne Encephalitis in Europe. Viruses. Mar 20 2023;15(3). doi:10.3390/v15030791
- Kohlmaier B, Schweintzger NA, Sagmeister MG, et al. Clinical Characteristics of Patients with Tick-Borne Encephalitis (TBE): A European Multicentre Study from 2010 to 2017. Microorganisms. Jun 30 2021;9(7). doi:10.3390/microorganisms9071420
- Kvam KA, Stahl JP, Chow FC, et al. Outcome and Sequelae of Autoimmune Encephalitis. J Clin Neurol. Jan 2024;20(1):3-22. doi:10.3988/jcn.2023.0242
- Hills S, Gould C, Cossaboom c. Tick-Borne Encephalitis. In: Nemhauser JB, ed. CDC Yellow Book 2024 – Health Information for International Travel. Oxford University Press; 2024.
- Vilibic-Cavlek T, Krcmar S, Bogdanic M, et al. An Overview of Tick-Borne Encephalitis Epidemiology in Endemic Regions of Continental Croatia, 2017-2023. Microorganisms. Feb 13 2024;12(2). doi:10.3390/microorganisms12020386
- Steininger P, Ensser A, Knoll A, Korn K. Results of Tick-Borne Encephalitis Virus (TBEV) Diagnostics in an Endemic Area in Southern Germany, 2007 to 2022. Viruses. Nov 30 2023;15(12). doi:10.3390/v15122357
- Czupryna P, Moniuszko A, Pancewicz SA, Grygorczuk S, Kondrusik M, Zajkowska J. Tick-borne encephalitis in Poland in years 1993-2008–epidemiology and clinical presentation. A retrospective study of 687 patients. Eur J Neurol. May 2011;18(5):673-9. doi:10.1111/j.1468-1331.2010.03278.x
- Jelenik Z, Keller M, Briggs B, et al. Tick-borne encephalitis and golden agers: position paper of the International Scientific Working Group on Tick-borne encephalitis (ISW-TBE). Wien Med Wochenschr. May 2010;160(9-10):247-51. doi:10.1007/s10354-010-0758-5
- Santonja I, Stiasny K, Essl A, Heinz FX, Kundi M, Holzmann H. Tick-Borne Encephalitis in Vaccinated Patients: A Retrospective Case-Control Study and Analysis of Vaccination Field Effectiveness in Austria From 2000 to 2018. J Infect Dis. Feb 14 2023;227(4):512-521. doi:10.1093/infdis/jiac075
- Radzisauskiene D, Urboniene J, Kaubrys G, et al. The epidemiology, clinical presentation, and predictors of severe Tick-borne encephalitis in Lithuania, a highly endemic country: A retrospective study of 1040 patients. PLoS One. 2020;15(11):e0241587. doi:10.1371/journal.pone.0241587
- Bogovic P, Lotric-Furlan S, Avsic-Zupanc T, Lusa L, Strle F. Factors associated with severity of tick-borne encephalitis: A prospective observational study. Travel Med Infect Dis. Nov-Dec 2018;26:25-31. doi:10.1016/j.tmaid.2018.10.003
- Nygren TM, Pilic A, Bohmer MM, Wagner-Wiening C, Wichmann O, Hellenbrand W. Recovery and sequelae in 523 adults and children with tick-borne encephalitis in Germany. Infection. Oct 2023;51(5):1503-1511. doi:10.1007/s15010-023-02023-w
- Lenhard T, Ott D, Jakob NJ, et al. Predictors, Neuroimaging Characteristics and Long-Term Outcome of Severe European Tick-Borne Encephalitis: A Prospective Cohort Study. PLoS One. 2016;11(4):e0154143. doi:10.1371/journal.pone.0154143
- Czarnowska A, Groth M, Okrzeja J, et al. A fatal case of tick-borne encephalitis in an immunocompromised patient: case report from Northeastern Poland and review of literature. Ticks Tick Borne Dis. Jan 2024;15(1):102273. doi:10.1016/j.ttbdis.2023.102273
- Lipowski D, Popiel M, Perlejewski K, et al. A Cluster of Fatal Tick-borne Encephalitis Virus Infection in Organ Transplant Setting. J Infect Dis. Mar 15 2017;215(6):896-901. doi:10.1093/infdis/jix040
- Wagner JN, Sonnberger M, Troescher A, et al. Patients with breakthrough tick-borne encephalitis suffer a more severe clinical course and display extensive magnetic resonance imaging changes. Eur J Neurol. Jul 2020;27(7):1201-1209. doi:10.1111/ene.14276
- Bogovic P, Lotric-Furlan S, Avsic-Zupanc T, et al. Low Virus-Specific IgG Antibodies in Adverse Clinical Course and Outcome of Tick-Borne Encephalitis. Microorganisms. Feb 7 2021;9(2)doi:10.3390/microorganisms9020332
- Carlstromer Berthen N, Tompa E, Olausson S, et al. The AxBioTick Study: Borrelia Species and Tick-Borne Encephalitis Virus in Ticks, and Clinical Responses in Tick-Bitten Individuals on the Aland Islands, Finland. Microorganisms. Apr 22 2023;11(5)doi:10.3390/microorganisms11051100
- Moniuszko A, Dunaj J, Swiecicka I, et al. Co-infections with Borrelia species, Anaplasma phagocytophilum and Babesia spp. in patients with tick-borne encephalitis. Eur J Clin Microbiol Infect Dis. Oct 2014;33(10):1835-41. doi:10.1007/s10096-014-2134-7
- Palus M, Vojtiskova J, Salat J, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. Jun 27 2013;10:77. doi:10.1186/1742-2094-10-77
- Faivre N, Verollet C, Dumas F. The chemokine receptor CCR5: multi-faceted hook for HIV-1. Retrovirology. Jan 23 2024;21(1):2. doi:10.1186/s12977-024-00634-1
- Kindberg E, Mickiene A, Ax C, et al. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis. Jan 15 2008;197(2):266-9. doi:10.1086/524709
- Mickiene A, Pakalniene J, Nordgren J, et al. Polymorphisms in chemokine receptor 5 and Toll-like receptor 3 genes are risk factors for clinical tick-borne encephalitis in the Lithuanian population. PLoS One. 2014;9(9):e106798. doi:10.1371/journal.pone.0106798
- Grygorczuk S, Osada J, Parczewski M, et al. The expression of the chemokine receptor CCR5 in tick-borne encephalitis. J Neuroinflammation. Feb 22 2016;13:45. doi:10.1186/s12974-016-0511-0
- Grygorczuk S, Dunaj-Malyszko J, Sulik A, et al. The Lack of the Association of the CCR5 Genotype with the Clinical Presentation and Frequency of Tick-Borne Encephalitis in the Polish Population. Pathogens. Mar 4 2022;11(3)doi:10.3390/pathogens11030318
- Barkhash AV, Perelygin AA, Babenko VN, et al. Variability in the 2′-5′-oligoadenylate synthetase gene cluster is associated with human predisposition to tick-borne encephalitis virus-induced disease. J Infect Dis. Dec 15 2010;202(12):1813-8. doi:10.1086/657418
- Barkhash AV, Babenko VN, Kobzev VF, Romaschenko AG, Voevoda MI. Polymorphism of 2′-5′-oligoadenylate synthetase (OAS) genes, associated with predisposition to severe forms of tick-borne encephalitis, in human populations of North Eurasia. Mol Biol. 2010;44(6):875-882. doi:10.1134/S002689331006004X
- Barkhash AV, Perelygin AA, Babenko VN, Brinton MA, Voevoda MI. Single nucleotide polymorphism in the promoter region of the CD209 gene is associated with human predisposition to severe forms of tick-borne encephalitis. Antiviral Res. Jan 2012;93(1):64-8. doi:10.1016/j.antiviral.2011.10.017
- Fortova A, Barkhash AV, Pychova M, et al. Genetic polymorphisms in innate immunity genes influence predisposition to tick-borne encephalitis. J Neurovirol. Dec 2023;29(6):699-705. doi:10.1007/s13365-023-01182-8
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. Sep 17 2009;461(7262):399-401. doi:10.1038/nature08309
- Barkhash AV, Babenko VN, Voevoda MI, Romaschenko AG. Association of IL28B and IL10 gene polymorphism with predisposition to tick-borne encephalitis in a Russian population. Ticks Tick Borne Dis. Jul 2016;7(5):808-812. doi:10.1016/j.ttbdis.2016.03.019
- Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. Oct 2005;79(20):12893-904. doi:10.1128/JVI.79.20.12893-12904.2005
- Labuda M, Randolph SE. Survival strategy of tick-borne encephalitis virus: cellular basis and environmental determinants. Zentralbl Bakteriol. Dec 1999;289(5-7):513-24. doi:10.1016/s0934-8840(99)80005-x
- Barkhash AV, Yurchenko AA, Yudin NS, et al. A matrix metalloproteinase 9 (MMP9) gene single nucleotide polymorphism is associated with predisposition to tick-borne encephalitis virus-induced severe central nervous system disease. Ticks Tick Borne Dis. May 2018;9(4):763-767. doi:10.1016/j.ttbdis.2018.02.010
- Czupryna P, Parczewski M, Grygorczuk S, et al. Analysis of the relationship between single nucleotide polymorphism of the CD209, IL-10, IL-28 and CCR5 D32 genes with the human predisposition to developing tick-borne encephalitis. Postepy Hig Med Dosw (Online). Jan 4 2017;71(1):788-796. doi:10.5604/01.3001.0010.3856
- Bogovic P, Kastrin A, Lotric-Furlan S, et al. Clinical and Laboratory Characteristics and Outcome of Illness Caused by Tick-Borne Encephalitis Virus without Central Nervous System Involvement. Emerg Infect Dis. Feb 2022;28(2):291-301. doi:10.3201/eid2802.211661
- Taba P, Schmutzhard E, Forsberg P, et al. EAN consensus review on prevention, diagnosis and management of tick-borne encephalitis. Eur J Neurol. Oct 2017;24(10):1214-e61. doi:10.1111/ene.13356
- ECDC. Factsheet. Tick-borne encephalitis. . Annual epidemiological report for 2018. 2019.
- Bogovic P, Lusa L, Korva M, et al. Inflammatory Immune Responses in the Pathogenesis of Tick-Borne Encephalitis. J Clin Med. May 22 2019;8(5)doi:10.3390/jcm8050731
- Ruzek D, Dobler G, Donoso Mantke O. Tick-borne encephalitis: pathogenesis and clinical implications. Travel Med Infect Dis. Jul 2010;8(4):223-32. doi:10.1016/j.tmaid.2010.06.004
- Mandl CW. Steps of the tick-borne encephalitis virus replication cycle that affect neuropathogenesis. Virus Res. Aug 2005;111(2):161-74. doi:10.1016/j.virusres.2005.04.007
- Palus M, Vancova M, Sirmarova J, Elsterova J, Perner J, Ruzek D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology. Jul 2017;507:110-122. doi:10.1016/j.virol.2017.04.012
- Ruzek D, Avsic Zupanc T, Borde J, et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antiviral Res. Apr 2019;164:23-51. doi:10.1016/j.antiviral.2019.01.014
- Blom K, Cuapio A, Sandberg JT, et al. Cell-Mediated Immune Responses and Immunopathogenesis of Human Tick-Borne Encephalitis Virus-Infection. Front Immunol. 2018;9:2174. doi:10.3389/fimmu.2018.02174
- Gustafson R, Forsgren M, Gardulf A, Granstrom M, Svenungsson B. Clinical manifestations and antibody prevalence of Lyme borreliosis and tick-borne encephalitis in Sweden: a study in five endemic areas close to Stockholm. Scand J Infect Dis. 1993;25(5):595-603. doi:10.3109/00365549309008548
- Gritsun TS, Lashkevich VA, Gould EA. Tick-borne encephalitis. Antiviral Res. Jan 2003;57(1-2):129-46. doi:10.1016/s0166-3542(02)00206-1
- Kaiser R. Tick-borne encephalitis. Infect Dis Clin North Am. Sep 2008;22(3):561-75, x. doi:10.1016/j.idc.2008.03.013
- Holzmann H, Aberle SW, Stiasny K, et al. Tick-borne encephalitis from eating goat cheese in a mountain region of Austria. Emerg Infect Dis. Oct 2009;15(10):1671-3. doi:10.3201/eid1510.090743
- Hudopisk N, Korva M, Janet E, et al. Tick-borne encephalitis associated with consumption of raw goat milk, Slovenia, 2012. Emerg Infect Dis. May 2013;19(5):806-8. doi:10.3201/eid1905.121442
- Kaiser R. The clinical and epidemiological profile of tick-borne encephalitis in southern Germany 1994-98: a prospective study of 656 patients. Brain. Nov 1999;122 ( Pt 11):2067-78. doi:10.1093/brain/122.11.2067
- Mickiene A, Laiskonis A, Gunther G, Vene S, Lundkvist A, Lindquist L. Tickborne encephalitis in an area of high endemicity in lithuania: disease severity and long-term prognosis. Clin Infect Dis. Sep 15 2002;35(6):650-8. doi:10.1086/342059
- Bogovic P, Strle F. Tick-borne encephalitis: A review of epidemiology, clinical characteristics, and management. World J Clin Cases. May 16 2015;3(5):430-41. doi:10.12998/wjcc.v3.i5.430
- Dumpis U, Crook D, Oksi J. Tick-borne encephalitis. Clin Infect Dis. Apr 1999;28(4):882-90. doi:10.1086/515195
- Kunz C. Tick-borne encephalitis in Europe. Acta Leiden. 1992;60(2):1-14.
- Granstrom M. Tick-borne zoonoses in Europe. Clin Microbiol Infect. Apr 1997;3(2):156-169. doi:10.1111/j.1469-0691.1997.tb00592.x
- Lotric-Furlan S, Petrovec M, Avsic-Zupanc T, Strle F. Clinical distinction between human granulocytic ehrlichiosis and the initial phase of tick-borne encephalitis. J Infect. Jan 2000;40(1):55-8. doi:10.1053/jinf.1999.0587
- Schultze D, Dollenmaier G, Rohner A, Guidi T, Cassinotti P. Benefit of detecting tick-borne encephalitis viremia in the first phase of illness. J Clin Virol. Feb 2007;38(2):172-5. doi:10.1016/j.jcv.2006.11.008
- Gunther G, Haglund M, Lindquist L, Forsgren M, Skoldenberg B. Tick-bone encephalitis in Sweden in relation to aseptic meningo-encephalitis of other etiology: a prospective study of clinical course and outcome. J Neurol. Apr 1997;244(4):230-8. doi:10.1007/s004150050077
- Lotric-Furlan S, Avsic-Zupanc T, Strle F. Is an isolated initial phase of a tick-borne encephalitis a common event? Clin Infect Dis. Jun 2000;30(6):987-8. doi:10.1086/313838
- Lotric-Furlan S, Avsic-Zupanc T, Strle F. An abortive form of tick-borne encephalitis (TBE)–a rare clinical manifestation of infection with TBE virus. Wien Klin Wochenschr. Jul 31 2002;114(13-14):627-9.
- Ustinova O, Volechova GM, Deviatkov MI, Gusmanova AI. [The clinico-epidemiological characteristics of tick-borne encephalitis in Perm Province]. Zh Mikrobiol Epidemiol Immunobiol. May-Jun 1997;(3):33-6. Kliniko-epidemiologicheskie osobennosti kleshchevogo entsefalita v Permskoi oblasti.
- Meyer PM, Zimmermann H, Goetschel P. Tick-borne encephalitis presenting as fever without localising signs–a case series. Eur J Pediatr. Jun 2010;169(6):767-9. doi:10.1007/s00431-009-1097-7
- Misic-Majerus L, Bujic N, Madaric V, Avsic-Zupanc T. [An abortive type of tick-borne meningoencephalitis]. Acta Med Croatica. 2003;57(2):111-6. Abortivni oblik krpeljnog meningoencefalitisa.
- Barp N, Trentini A, Di Nuzzo M, Mondardini V, Francavilla E, Contini C. Clinical and laboratory findings in tick-borne encephalitis virus infection. Parasite Epidemiol Control. Aug 2020;10:e00160. doi:10.1016/j.parepi.2020.e00160
- Misic Majerus L, Dakovic Rode O, Ruzic Sabljic E. [Post-encephalitic syndrome in patients with tick-borne encephalitis]. Acta Med Croatica. Oct 2009;63(4):269-78. Postencefaliticki sindrom u bolesnika s krpeljnim meningoencefalitisom.
- Logar M, Bogovic P, Cerar D, Avsic-Zupanc T, Strle F. Tick-borne encephalitis in Slovenia from 2000 to 2004: comparison of the course in adult and elderly patients. Wien Klin Wochenschr. Nov 2006;118(21-22):702-7. doi:10.1007/s00508-006-0699-6
- Kaiser R. Tick-borne encephalitis (TBE) in Germany and clinical course of the disease. Int J Med Microbiol. Jun 2002;291 Suppl 33:58-61. doi:10.1016/s1438-4221(02)80012-1
- Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet. May 31 2008;371(9627):1861-71. doi:10.1016/S0140-6736(08)60800-4
- Karelis G, Bormane A, Logina I, et al. Tick-borne encephalitis in Latvia 1973-2009: epidemiology, clinical features and sequelae. Eur J Neurol. Jan 2012;19(1):62-8. doi:10.1111/j.1468-1331.2011.03434.x
- Jereb M, Muzlovic I, Avsic-Zupanc T, Karner P. Severe tick-borne encephalitis in Slovenia: epidemiological, clinical and laboratory findings. Wien Klin Wochenschr. Jul 31 2002;114(13-14):623-6.
- Jereb M, Karner P, Muzlovic I, Jurca T. Severe tick-borne encephalitis in Slovenia in the years 2001-2005: time for a mass vaccination campaign? Wien Klin Wochenschr. Dec 2006;118(23-24):765-8. doi:10.1007/s00508-006-0728-5
- Bogovic P, Lotric-Furlan S, Avsic-Zupanc T, et al. Comparison of Clinical, Laboratory and Immune Characteristics of the Monophasic and Biphasic Course of Tick-Borne Encephalitis. Microorganisms. Apr 10 2021;9(4)doi:10.3390/microorganisms9040796
- Bannova GG, Sarmanova ES, Karavanov AS, Bychkova MV, Pivanova GP. [Biological properties of tick-borne encephalitis strains isolated in different parts of its geographic range]. Vopr Virusol. Jan-Feb 1982;(1):41-5. Izuchenie biologicheskikh svoistv shtammov virusa kleshchevogo entsefalita, vydelennykh v raznykh chastiakh ego areala.
- Dekonenko EP, Umanskii KG. [Sequelae of different clinical forms of the acute stage of tick-borne encephalitis]. Zh Nevropatol Psikhiatr Im S S Korsakova. 1984;84(2):202-7. Posledstviia razlichnykh klinicheskikh form ostrogo perioda kleshchevogo entsefalita.
- Enzinger C, Melisch B, Reischl A, Simbrunner J, Fazekas F. Polyradiculitis as a predominant symptom of tick-borne encephalitis virus infection. Arch Neurol. Jul 2009;66(7):904-5. doi:10.1001/archneurol.2009.117
- Lotric-Furlan S, Strle F. Peripheral facial palsy in patients with tick-borne encephalitis. Clin Microbiol Infect. Oct 2012;18(10):1027-32. doi:10.1111/j.1469-0691.2011.03719.x
- Kleiter I, Steinbrecher A, Flugel D, Bogdahn U, Schulte-Mattler W. Autonomic involvement in tick-borne encephalitis (TBE): report of five cases. Eur J Med Res. Jun 30 2006;11(6):261-5.
- Neumann B, Schulte-Mattler W, Brix S, et al. Autonomic and peripheral nervous system function in acute tick-borne encephalitis. Brain Behav. Aug 2016;6(8):e00485. doi:10.1002/brb3.485
- Poschl P, Kleiter I, Grubwinkler S, et al. [Severe tick-borne encephalomyelitis with lack of cerebrospinal fluid pleocytosis]. Fortschr Neurol Psychiatr. Oct 2009;77(10):591-3. Schwere Fruhsommer-Meningo-Enzephalomyelitis ohne Liquor-Pleozytose. doi:10.1055/s-0028-1109768
- Stupica D, Strle F, Avsic-Zupanc T, Logar M, Pecavar B, Bajrovic FF. Tick borne encephalitis without cerebrospinal fluid pleocytosis. BMC Infect Dis. Nov 18 2014;14:614. doi:10.1186/s12879-014-0614-0
- Poponnikova TV. Specific clinical and epidemiological features of tick-borne encephalitis in Western Siberia. Int J Med Microbiol. May 2006;296 Suppl 40:59-62. doi:10.1016/j.ijmm.2006.01.023
- Mukhin KY, Mameniskiene R, Mironov MB, et al. Epilepsia partialis continua in tick-borne Russian spring-summer encephalitis. Acta Neurol Scand. May 2012;125(5):345-52. doi:10.1111/j.1600-0404.2011.01575.x
- Mansfield KL, Johnson N, Phipps LP, Stephenson JR, Fooks AR, Solomon T. Tick-borne encephalitis virus – a review of an emerging zoonosis. J Gen Virol. Aug 2009;90(Pt 8):1781-1794. doi:10.1099/vir.0.011437-0
- Sidorenko M, Radzijevskaja J, Mickevicius S, et al. Phylogenetic characterisation of tick-borne encephalitis virus from Lithuania. PLoS One. 2024;19(2):e0296472. doi:10.1371/journal.pone.0296472
- Kaiser R. [Epidemiology and progress of early summer meningoencephalitis in Baden-Wurttemberg between 1994 and 1999. A prospective study of 731 patients]. Dtsch Med Wochenschr. Sep 29 2000;125(39):1147-53. Epidemiologie und Verlauf der Fruhsommer-Meningoenzephalitis in Baden-Wurttemberg zwischen 1994 und 1999. Eine prospektive Studie an 731 Patienten. doi:10.1055/s-2000-7668
- Grygorczuk S, Mierzynska D, Zdrodowska A, et al. Tick-borne encephalitis in north-eastern Poland in 1997-2001: a retrospective study. Scand J Infect Dis. 2002;34(12):904-9. doi:10.1080/0036554021000026979
- Bogovic P, Kastrin A, Lotric-Furlan S, et al. Comparison of laboratory and immune characteristics of the initial and second phase of tick-borne encephalitis. Emerg Microbes Infect. Dec 2022;11(1):1647-1656. doi:10.1080/22221751.2022.2086070
- Lotric-Furlan S, Strle F. Thrombocytopenia–a common finding in the initial phase of tick-borne encephalitis. Infection. Jul-Aug 1995;23(4):203-6. doi:10.1007/BF01781197
- Bogovic P, Lotric-Furlan S, Ogrinc K, et al. Elevated levels of serum muscle enzymes in the initial phase of tick-borne encephalitis. Infect Dis (Lond). Jun 2024;56(6):504-509. doi:10.1080/23744235.2024.2335349
- Condos AM, Wangaryattawanich P, Rath TJ. Bacterial, Viral, and Prion Infectious Diseases of the Brain. Magn Reson Imaging Clin N Am. May 2024;32(2):289-311. doi:10.1016/j.mric.2023.11.001
- Fjordside L, Nissen MS, Florescu AM, et al. Validation of a risk score to differentiate autoimmune and viral encephalitis: a Nationwide Cohort Study in Denmark. J Neurol. May 18 2024;doi:10.1007/s00415-024-12392-3
- Mohan S, Jain KK, Arabi M, Shah GV. Imaging of meningitis and ventriculitis. Neuroimaging Clin N Am. Nov 2012;22(4):557-83. doi:10.1016/j.nic.2012.04.003
- Duong MT, Rudie JD, Mohan S. Neuroimaging Patterns of Intracranial Infections: Meningitis, Cerebritis, and Their Complications. Neuroimaging Clin N Am. Feb 2023;33(1):11-41. doi:10.1016/j.nic.2022.07.001
- Horger M, Beck R, Fenchel M, et al. Imaging findings in tick-borne encephalitis with differential diagnostic considerations. AJR Am J Roentgenol. Aug 2012;199(2):420-7. doi:10.2214/AJR.11.7911
- Saremi F, Helmy M, Farzin S, Zee CS, Go JL. MRI of cranial nerve enhancement. AJR Am J Roentgenol. Dec 2005;185(6):1487-97. doi:10.2214/AJR.04.1518
- Bloch KC, Glaser C, Gaston D, Venkatesan A. State of the Art: Acute Encephalitis. Clin Infect Dis. Sep 11 2023;77(5):e14-e33. doi:10.1093/cid/ciad306
- Pichler A, Sellner J, Harutyunyan G, et al. Magnetic resonance imaging and clinical findings in adults with tick-borne encephalitis. J Neurol Sci. Apr 15 2017;375:266-269. doi:10.1016/j.jns.2017.02.003
- Abbuehl LS, Branca M, Ungureanu A, et al. Magnetic resonance imaging in acute meningoencephalitis of viral and unknown origin: frequent findings and prognostic potential. Front Neurol. 2024;15:1359437. doi:10.3389/fneur.2024.1359437
- Dietmann A, Putzer D, Beer R, et al. Cerebral glucose hypometabolism in Tick-Borne Encephalitis, a pilot study in 10 Patients. Int J Infect Dis. Oct 2016;51:73-77. doi:10.1016/j.ijid.2016.06.022
- Czarnowska A, Kapica-Topczewska K, Garkowski A, et al. Severe tick-borne encephalitis in a patient recovered from COVID 19. Ticks Tick Borne Dis. Jul 2022;13(4):101940. doi:10.1016/j.ttbdis.2022.101940
- Czupryna P, Tarasow E, Moniuszko-Malinowska A, et al. MRI and planimetric CT follow-up study of patients with severe tick-borne encephalitis. Infect Dis (Lond). 2016;48(1):74-81. doi:10.3109/23744235.2015.1083119
- Neill L, Checkley AM, Benjamin LA, et al. Rhombencephalitis and Myeloradiculitis Caused by a European Subtype of Tick-Borne Encephalitis Virus. Emerg Infect Dis. Dec 2019;25(12):2317-2319. doi:10.3201/eid2512.191017
- Schellinger PD, Schmutzhard E, Fiebach JB, Pfausler B, Maier H, Schwab S. Poliomyelitic-like illness in central European encephalitis. Neurology. Jul 25 2000;55(2):299-302. doi:10.1212/wnl.55.2.299
- Feige J, Moser T, Hauer L, Pikija S, Sellner J. Clinical Challenges in a 49-Year-Old Patient with Severe Tick-Borne Myeloradiculitis Despite Complete Active Vaccination. Vaccines (Basel). Feb 20 2020;8(1)doi:10.3390/vaccines8010093
- Racz A, Schaller G, Lunkenheimer J, et al. Isolated meningomyeloradiculitis following infection with tick borne encephalitis virus. Clin Neurol Neurosurg. Nov 2012;114(9):1263-5. doi:10.1016/j.clineuro.2012.02.047
- Sellner J, Trinka E. Epilepsy associated with viral encephalitis. In: Shorvon S, Guerrini R, Schachter S, Trinka E, eds. The causes of epilepsy. Cambridge University Press; 2019.
- Schmolck H, Maritz E, Kletzin I, Korinthenberg R. Neurologic, neuropsychologic, and electroencephalographic findings after European tick-borne encephalitis in children. J Child Neurol. Jun 2005;20(6):500-8. doi:10.1177/088307380502000606
- Sipila JOT. Adult-onset encephalitis over two decades in easternmost Finland. Neuroepidemiology. Feb 28 2024;doi:10.1159/000538020
- Zelano J, Westman G. Epilepsy after brain infection in adults: A register-based population-wide study. Neurology. Dec 15 2020;95(24):e3213-e3220. doi:10.1212/WNL.0000000000010954
- Varnaite R, Gredmark-Russ S, Klingstrom J. Deaths from Tick-Borne Encephalitis, Sweden. Emerg Infect Dis. Jul 2022;28(7):1471-1474. doi:10.3201/eid2807.220010
- Beaute J, Spiteri G, Warns-Petit E, Zeller H. Tick-borne encephalitis in Europe, 2012 to 2016. Euro Surveill. Nov 2018;23(45)doi:10.2807/1560-7917.ES.2018.23.45.1800201
- Slunge D, Boman A, Studahl M. Burden of Tick-Borne Encephalitis, Sweden. Emerg Infect Dis. Feb 2022;28(2):314-322. doi:10.3201/eid2802.204324
- Bogovic P, Stupica D, Rojko T, et al. The long-term outcome of tick-borne encephalitis in Central Europe. Ticks Tick Borne Dis. Feb 2018;9(2):369-378. doi:10.1016/j.ttbdis.2017.12.001
- Griska V, Pranckeviciene A, Pakalniene J, et al. Long-term neurological and neurocognitive impairments after tick-borne encephalitis in Lithuania – a prospective study. Infect Dis (Lond). May 6 2024:1-11. doi:10.1080/23744235.2024.2346793
- Eyer L, Seley-Radtke K, Ruzek D. New directions in the experimental therapy of tick-borne encephalitis. Antiviral Res. Feb 2023;210:105504. doi:10.1016/j.antiviral.2022.105504
- Czupryna P, Moniuszko A, Garkowski A, Pancewicz S, Zajkowska J. Comparison of hyponatremia and SIADH frequency in patients with tick borne encephalitis and meningitis of other origin. Scand J Clin Lab Invest. 2016;76(2):159-64. doi:10.3109/00365513.2015.1129669
- Sonneville R, Jaquet P, Vellieux G, de Montmollin E, Visseaux B. Intensive care management of patients with viral encephalitis. Rev Neurol (Paris). Jan-Feb 2022;178(1-2):48-56. doi:10.1016/j.neurol.2021.12.002