
Chapter 2a:
Virology
Daniel Růžek, Kentaro Yoshii, Marshall E. Bloom and Ernest A. Gould
Key Points
- TBEV is the most medically important member of the tick-borne serocomplex group within
the genus Flavivirus, family Flaviviridae. - Three antigenic subtypes of TBEV correspond to the 3 recognized genotypes: European
(TBEV-EU), also known as Western, Far Eastern (TBEV-FE), and Siberian (TBEV-SIB).
An additional 2 genotypes have been identified in the Irkutsk region of Russia, currently named
TBE virus Baikalian subtype (TBEV-BKL) and TBE virus Himalayan subtype (Himalayan and “178-79” group; TBEV-HIM). - TBEV virions are small enveloped spherical particles about 50 nm in diameter.
- The TBEV genome consists of a single-stranded positive sense RNA molecule.
- The genome encodes one open reading frame (ORF), which is flanked by untranslated
(non-coding) regions (UTRs). - The 5′-UTR end has a methylated nucleotide cap for canonical cellular translation. The 3′-UTR is not polyadenylated and is characterized by extensive length and sequence heterogeneity.
- The ORF encodes one large polyprotein, which is co- and post-translationally cleaved into 3 structural proteins (C, prM, and E) and 7 non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5).
- TBEV replicates in the cytoplasm of the host cell in close association with virus-induced intracellular membrane structures. Virus assembly occurs in the endoplasmic reticulum.
The immature virions are transported to the Golgi complex, and mature virions pass through the host secretory pathway and are finally released from the host cell by fusion of the transport vesicle membrane with the plasma membrane.
Virus Classification
Tick-borne encephalitis virus (TBEV) is the most medically important member of the tick-borne serocomplex group within the genus Flavivirus, family Flaviviridae (from the Latin flavus – ‘yellow’, referring to the prototype virus, yellow fever virus).
The genus Flavivirus comprises over 70 virus species, many of which are important human pathogens.1 Besides TBEV, these include mosquito-borne viruses such as dengue viruses, Japanese encephalitis virus, yellow fever virus, Zika virus, and many others. Virtually the entire human population lives where at least one flavivirus species is endemic.1 Moreover, many flaviviruses have recently expanded their endemic areas, being introduced to novel loci either on new continents (West Nile virus, Zika virus, etc.) or to areas with higher altitude or latitude (TBEV as an example).2-3 For these reasons, flaviviruses pose an important threat to public and animal health. Moreover, they have high zoonotic potential because they can infect a broad range of hosts and vectors including domestic animals.
Most of the known flaviviruses are transmitted horizontally between hematophagous arthropods (ticks or mosquitoes) and their vertebrate hosts. They are therefore considered to be dual-host viruses. Depending on the recognized arthropod vector, they are divided into mosquito-borne or tick-borne viruses.
The term ‘arbovirus’ (an acronym from ‘arthropod-borne virus’) is non-taxonomic but is frequently used for viruses that cycle between vertebrates and arthropod vectors. However, not all flaviviruses are arboviruses – some are vertebrate-specific (also called ‘No known vector’ and further divided into rodent-specific and bat-specific flaviviruses)4 while some are insect-specific.5 These classifications reflect the adaptation of the viruses to particular invertebrate or vertebrate hosts, and modes of virus transmission in nature.
Tick-borne flaviviruses (TBFVs) are further divided into mammalian and seabird TBFVs. While the seabird TBFV are non-pathogenic for humans, mammalian TBFV include several important human pathogens; in particular, TBEV, Kyasanur Forest disease virus (KFDV), Omsk hemorrhagic fever virus (OHFV), Powassan/Deer tick virus (POWV), and louping ill virus (LIV), which together with Langat virus (LGTV), for which there are no known cases of natural human disease, comprise a group known as the ‘TBEV serocomplex’ (Figure 1). All TBFVs are closely related antigenically and antibodies against one TBFV often cross-react with the other TBFVs, which should be taken into consideration when interpreting serological tests in areas where more than one TBFV co-circulates. The broadest cross-reactivity is seen in hemagglutination inhibition assays, whereas the highest specificity is seen in neutralization assays.6
Figure 1: TBEV phylogenetic tree
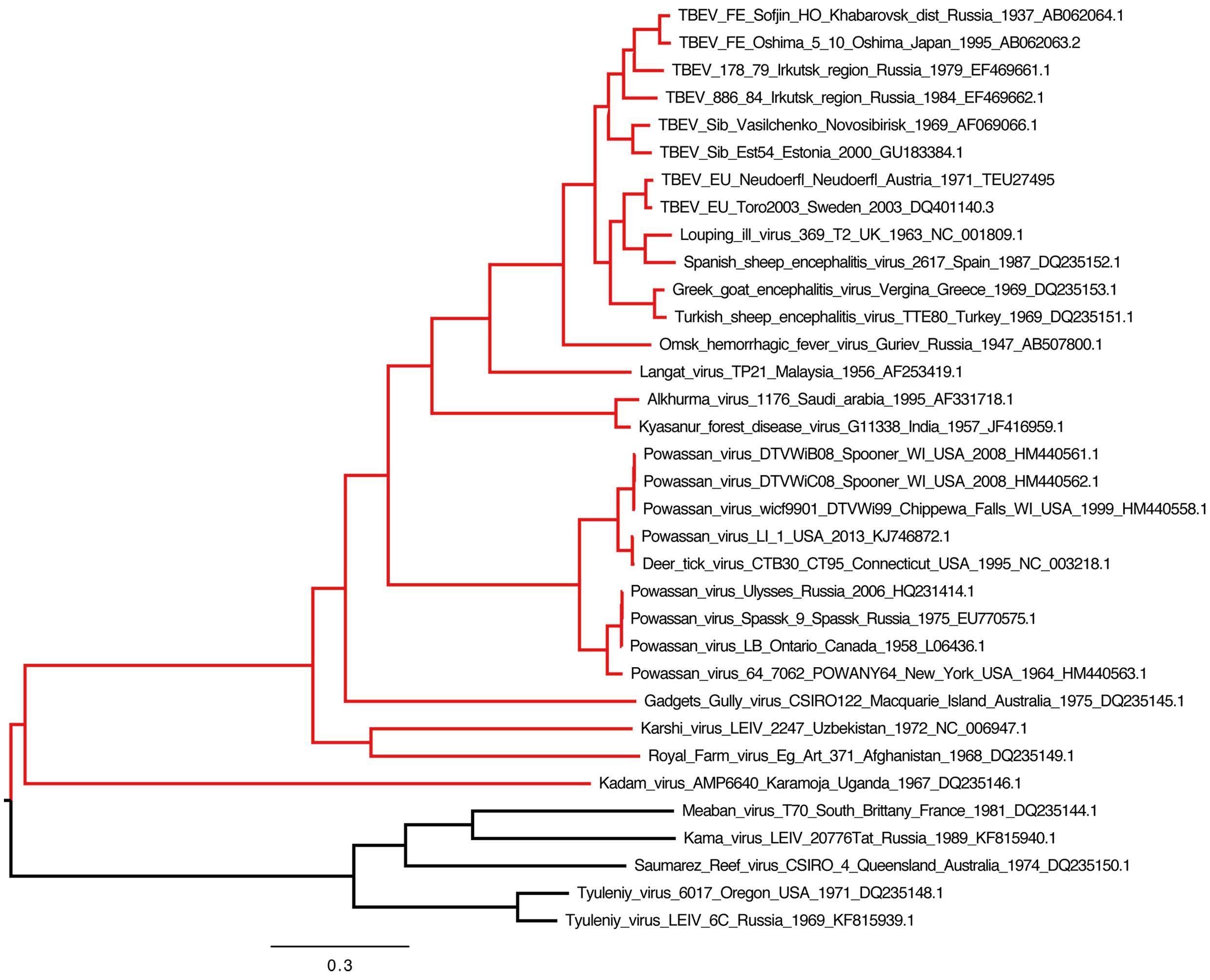
Click the image above to enlarge
Phylogenetic tree illustrating the relationships between representative members of the TBEV complex (highlighted in red). Complete genome open reading frame sequences were retrieved from genbank and aligned using the gins option in mafft v7.266. The tree was constructed with RAxML v.8.2.9 using the GTR+G model of nucleotide evolution and 1000 bootstrap replicates. The resulting tree was visualized and edited in Figtree v.1.4.1. All branches have maximum bootstrap support (not shown). The tree was midpoint rooted for visual purposes only. The lowest clade (black) contains members of the divergent seabird tick-associated virus complex (Meaban virus through Tyuleniy virus). We gratefully acknowledge the assistance of Dr John Pettersson (Zoonosis Science Center, Uppsala University, Sweden) who prepared and supplied the tree.
Although all TBFVs are closely related genetically and antigenically, they cause diverse clinical manifestations in humans: OHFV and KFDV (including a subtype of this virus, Alkhurma hemorrhagic fever virus) induce hemorrhagic fever syndromes, while the others cause neurological disease. Importantly, the hemorrhagic fever associated TBFVs and encephalitogenic TBFVs do not form separate phylogenetic lineages and no specific determinants in the genomes of these viruses have been associated with particular disease manifestations.7,8
Three main antigenic subtypes of TBEV correspond to the 3 recognized genotypes: Western, also known as European (TBEV-EU; previously Central European encephalitis; prototype strain Neudoerfl), Far Eastern (TBEV-FE; previously Russian spring-summer encephalitis; prototype strain Sofjin), and Siberian (TBEV-Sib; previously Western Siberian encephalitis; prototype strains Zausaev and Vasilchenko).10,11 Two additional lineages; i.e., “178-79” and “886-84 group”, named as Baikalian TBEV (TBEV-Bkl) respectively, have been identified in Eastern Siberia and proposed as TBEV subtypes.115,116 The geographical distribution and clinical significance of these newly identified genotypes remains to be determined. However, some studies indicate that 0.6-6% of TBEV strains circulating in Eastern Siberia might belong to these new genotypes.12 Another new potential TBEV subtype (Himalayan – TBEV-Him) was identified recently in wild rodents in Qinghai-Tibet Plateau in China.117
Comparison of the complete coding sequences of all recognized TBFV species led to a new taxonomic proposal, viz. the assignment of TBEV and LIV to a single species (TBEV) encompassing 4 viral types; i.e., Western TBEV (TBEV-EU); Eastern TBEV (TBEV-Sib and TBEV-FE); Turkish sheep TBEV, including Greek goat encephalitis virus subtype; and Louping ill TBEV, the latter having Spanish, British, and Irish subtypes.13 This classification was supported by the fact that, based on antigenic properties, the European TBEV strains are more closely related to LIV than to TBEV-FE and TBEV-Sib strains.14,15
All TBFVs are thought to have shared a common ancestor, which diverged from mosquito-borne flaviviruses in Africa less than 5000 years ago.16-18 However, some studies suggest that this split might have occurred as long as 50,000 years ago.19 The descendant TBFV species evolved and spread through Asia and then more recently westwards through Europe as they adapted to different host and tick species.16-18 In comparison with mosquito-borne flaviviruses, TBFVs evolved nearly twice as slowly, primarily due to the long life-cycle of the Ixodes tick vector.16,20,21 Overall, it was concluded that there is a direct correlation between genetic and geographic distance of individual TBFV species16,22 and, furthermore, that the evolution and dispersal of these viruses is relatively slower than that of the mosquito-transmitted viruses. In addition, the evolution is not significantly influenced by migratory birds or international trade.23
Virion structure and morphology
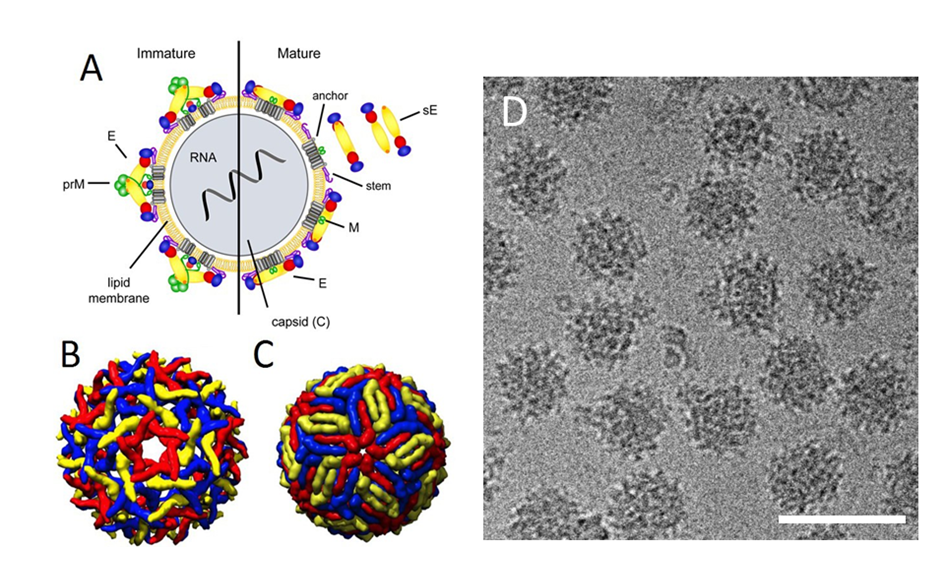
Infectious TBEV virions are small spherical particles about 50 nm in diameter with no obvious distinct projections. The mature virions contain an electron-dense core approximately 30 nm in diameter which is surrounded by a lipid bilayer (Figure 2).24 The nucleocapsid core consists of single-stranded positive-polarity genomic ribonucleic acid (RNA) molecule (11 kb) and the capsid protein C (12 kDa). The surface of the lipid membrane incorporates an envelope glycoprotein (E, 53K) and a membrane glycoprotein (M, 8K) (Figure 2).
Figure 2: TBEV particles

Click the image above to enlarge
Figures are reproduced from Füzik et al. Nat Commun. 2018 Jan 30;9(1):436. doi: 10.1038/s41467-018-02882-0 (https://www.nature.com/articles/s41467-018-02882-0) based on CC-BY 4.0 licence.
- Cryo-EM micrograph of TBEV particles. The sample contained mature, immature (white arrows), half-mature (white arrowheads), and damaged (black arrows) particles. Scalebar, 100 nm.
- B-factor sharpened electron-density map of TBEV virion, rainbow-colored according to distance from particle center. Scalebar, 10 nm.
- Molecular surface of TBEV virion low-pass filtered to 7 Å. The three E-protein subunits within each icosahedral asymmetric unit are shown in red, green, and blue. Scalebar, 10 nm.
- Central slice of TBEV electron density map perpendicular to the virus 5-fold axis. The virus membrane is deformed by the transmembrane helices of E-proteins and M-proteins. The lower right quadrant of the slice is color-coded as follows: nucleocapsid—blue; inner and outer membrane leaflets—orange; M-proteins—red; E-proteins—green. Scalebar, 10 nm.
The glycosylated E protein is also a major antigenic determinant of the virus and induces immune responses in infected mammalian hosts. It also contains the sites for virus binding to receptors on the surface of susceptible host cells and subsequent pH-mediated fusion of the viral E protein with endosomal membranes during entry of viral RNA into the cell.
In the mature infectious virions, the M protein has been proteolytically cleaved from the precursor (pr)M protein. This post-translational process occurs during the maturation of nascent viral particles within the secretory pathway and immediately before release of the infectious virions from the infected cell. In immature non-infectious particles, prM and E proteins form hetero-dimers and exist as trimers covering the virion surface. At this stage, the pr part of prM occludes the fusion domain of the E glycoprotein, preventing premature fusion with cell membranes within the secretory pathway (Figure 3).
Figure 3
Click the image above to enlarge
- Schematic model of a flavivirus particle. Left panel: immature virion, right panel: mature virion. The surface of immature particles consists of 60 spikes composed of trimers of prM-E heterodimers. Mature particles are formed after prM cleavage and contain 90 E homodimers. (From Vratskikh O, Stiasny K, Zlatkovic J, et al. Dissection of antibody specificities induced by yellow fever vaccination. PLoS Pathog 2013;9:e1003458. figshare: https://dx.doi.org/10.1371/journal.ppat.1003458.g001 (CC BY)).
- Pseudoatomic cryo-EM reconstruction model of the immature flavivirus particle (PDB: 2OF6).
- Pseudoatomic cryo-EM reconstruction model of the mature flavivirus particle (PDB: 3J0B).
- Cryo-EM micrograph of immature TBEV particles (kindly provided by Tibor Füzik and Pavel Plevka, with permission). Scalebar, 100 nm.
In the trans-Golgi compartment, the pr is cleaved from prM by a cell furin-like protease; this is followed by the conformational change, rotation, and rearrangement of E proteins from 60 antiparallel trimers into 90 anti-parallel dimers, forming an unusual ‘herring-bone’ pattern with icosahedral symmetry and resulting in the viral particles being mature and fully infectious. However, the efficiency of prM cleavage varies for different flaviviruses; cleavage is therefore not always absolute. Thus, immature particles may also be released as a proportion of the infectious/non-infectious virus pool.25
The structure of purified TBEV particles has recently been determined at near atomic resolution of 3.9 Å by reconstruction of cryo-electronmicroscopic images (Figure 2).118 The study revealed a relatively smooth outer surface of the particle, and E and M proteins organized in a similar manner to that in other flaviviruses. The surface of the TBEV virion is covered with small protrusions formed by glycans attached to the E-protein molecules.118 Both E-proteins and M-proteins are anchored in the virion membrane, each by two trans-membrane helices. Viral envelope membrane is not spherical; instead the shape of the membrane closely follows the inner surface of the protein envelope and is deformed by insertions of the trans-membrane helices of E-proteins and M-proteins.118
Recombinant sub-viral particles (RSPs) are of T-1 icosahedral symmetry formed by 30 E protein dimers. They have the same antigenic properties as wild-type virus. They can be used for vaccination purposes and represent an established model system for flavivirus membrane fusion because they have fusion characteristics similar to those of infectious virions.28
Viral genome
The nucleocapsid is formed from a single viral RNA genome and multiple copies of the C protein. The RNA binding domains of the C protein molecules are located at their N- and C-termini and are separated by hydrophobic regions. The nucleocapsid is less ordered and as for other flaviviruses, no discernible symmetry was detected in cryoelectron microscopic reconstructions.26 Instead, the C protein is arranged in a cage-like structure surrounding the viral genome. The icosahedral symmetry is, therefore, directed by surface proteins rather than by the nucleocapsid protein.
In addition to mature virions, smaller (approximately 14 nm in diameter) non-infectious particles are released from the infected cells. These particles lack nucleocapsid and consist of E and M proteins only; they are called sedimenting (70S) hemagglutinin (SHA).
Similar RSPs of a slightly larger size (approximately 30 nm in diameter) can be produced by cells expressing only prM and E proteins.27
The TBEV genome consists of a single-stranded positive sense RNA molecule, approximately 11 kilobases in length. The genome encodes 1 open reading frame (ORF) of over 10,000 bases, which is flanked by untranslated (non-coding) regions (UTRs). The ORF encodes 1 large polyprotein of approximately 3400 amino acids, which is co- and post-translationally cleaved by viral and cellular proteases into 3 structural proteins (C, prM, and E) and 7 non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5)29 (Figure 4). A second short upstream ORF is present in the 5′-UTR of some TBEV strains. However, no protein encoded by this ORF has been found in TBEV-infected cells, indicating that it is not expressed nor is present at undetectable concentrations, suggesting that this additional ORF has either minor or no biological role in the TBEV replication cycle.30 A common feature of all flavivirus genomes is their high purine content and low GC and UA doublet frequencies, which may influence translation of the genome and/or reflect the requirement for flaviviruses to grow in different hosts and cell types; however, a specific role for this unique genomic characteristic remains unclear.31 A replication enhancer element (REE) has been found within the capsid gene of TBEV. The REE folds as a long stable stem-loop (designated SL6), conserved among all TBFVs. Although SL6 REE is not essential for growth in tissue culture, it acts to up-regulate virus replication.32
In addition to coding for the polyprotein, the genome has RNA structural motifs that play a crucial role in the viral life-cycle.33 In particular, the untranslated regions form secondary stem-loop structures that probably serve as cis-acting elements for genome replication, translation, and/or packaging.33-36 The 5’-UTR contains a type 1 cap (m7GpppAmG), followed by a conserved stem-loop structure. The 3’-UTR is not polyadenylated and is characterized by extensive length and sequence heterogeneity.37 This region of the viral genome can be divided into 2 parts: a proximal (localized behind the ‘stop’ codon of the ORF) and a distal (‘core’, the 3′ terminus itself). The distal part of this region (approximately 340 nt) is highly conserved, whilst the proximal part is a noticeably variable segment with common deletions and insertions.34-36
Figure 4
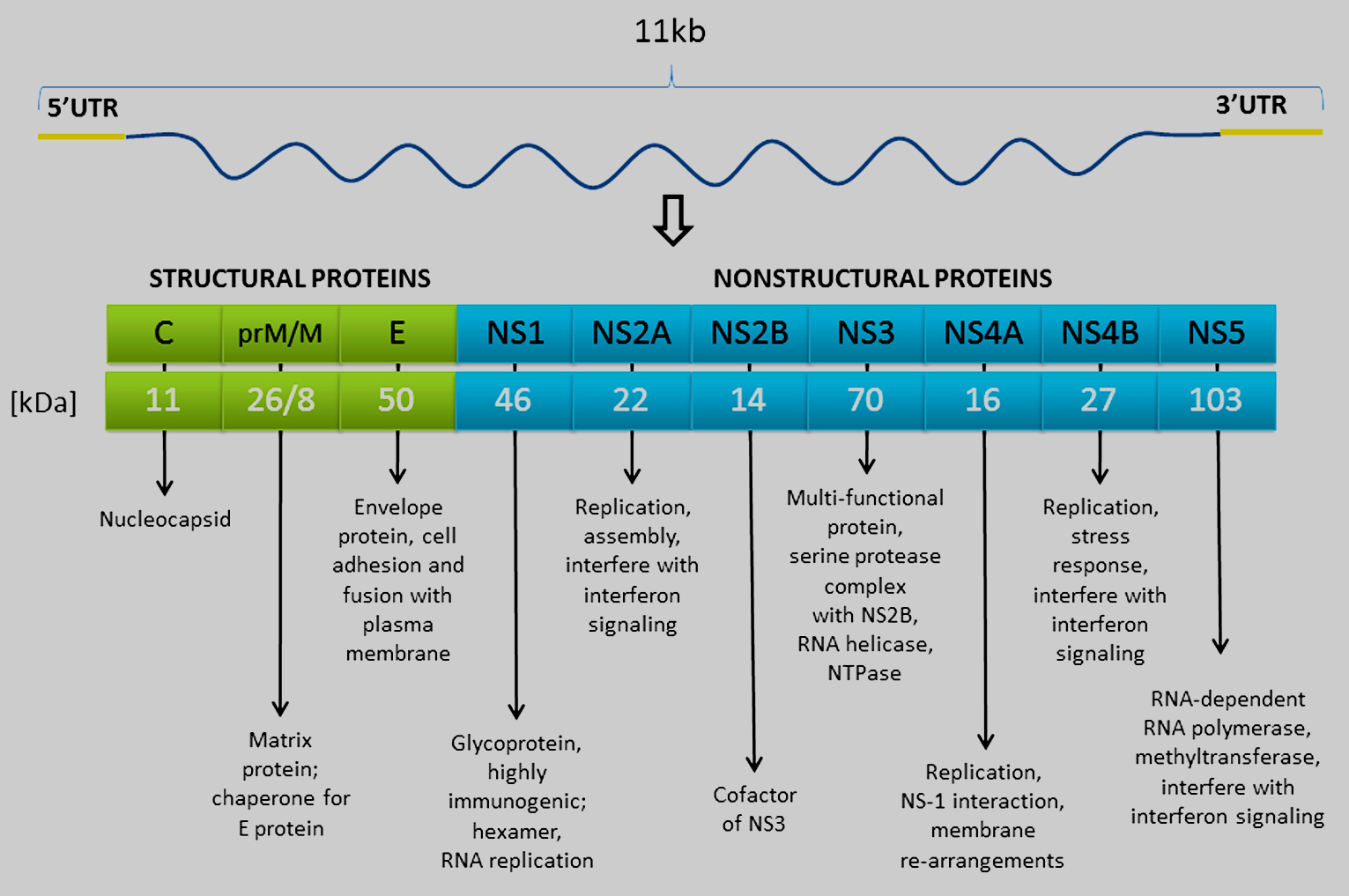
Click the image above to enlarge
Genome organization of TBEV and processing pathways of the polyprotein. A schematic representation of the TBEV genome with the 5′ and 3′ non-translated regions (NTRs) is shown in the top; the translation products are given below (kindly provided by Martin Palus, with permission).
RNA structural models demonstrate that flavivirus genomes, including TBFVs, form dsRNA cyclization stems or ‘panhandles’ at their 5′- and 3′-termini. The ‘panhandle’ of the TBFV group (5’CYCL) is formed by a perfectly conserved continuous 21-nucleotide sequence located in the 5′-UTR. The 5′-UTR and 3′-UTR sequences directly involved in cyclization are located downstream from the 5′ Y-shaped structure and the 3′ long stable hairpin, respectively. The terminal 5′-UTR and 3′-UTR regions not involved in cyclization also show homology, suggesting they are evolutionary remnants of a long cyclization domain that probably emerged through duplication of 1 of the UTR termini.38
5’-untranslated region
The 5’-UTR is 132 nucleotides long in most TBEV strains and its secondary structure is highly conserved among different TBEV strains.36 Common secondary structures in this region can also be found among different flaviviruses, although the sequence is diverse.31 The function of these conserved secondary structures is probably related to translation of the genome and in the complementary RNA strand serves as a site for initiation of synthesis of positive-stranded RNA molecules.39
The folding of 333 nt as a reverse complement of the 5′-end (3′-end of the negative-stranded RNA) of TBEV revealed a stem-loop pattern different from the 3′-UTR of positive-stranded RNA. However, 2 nucleotide regions in these 3′-ends are identical and conserved among all TBFVs. One of these, an 11-nt region, forms a loop within the folding pattern at the 3′-end of the negative strand and a stem at the 3′-UTR of the positive strand.34 These structural motifs at the 5′ and 3′-UTR termini could be recognition sites for viral RNA polymerase.34
The alignment of the 5′-UTRs of different TBFVs demonstrated an internal hypervariable domain in which Powassan virus has a deletion of 27 bases.34 The predicted folding of the 5′-UTR sequence produces a stem-loop structure similar for all TBFV, and the 27 nt deletion in the Powassan virus has no effect on the typical 5′-UTR folding.34 This indicates that the length of stem-loop structure 3 is not critical for virus infectivity.34
3’-untranslated region
The alignment of 3′-UTRs of all TBFVs revealed 2 nucleotide regions, 1 about 340 bases in length, of conserved sequence at the extreme 3′-end (designated C3′- UTR) and another hypervariable region placed between the stop codon and the C3′-UTR where even strains from a single species showed deletions of different lengths,34 whereas some TBEV strains have a 30-250 nt long poly(A) sequence in this region.37 Deletions or a poly(A) sequence insertion in the variable region were found in strains passaged in mammalian cell culture,40 and deletions of different lengths were also observed in TBEV strains isolated from human patients.41-43 It was suggested that the hypervariable region could act as a spacer separating the folded 3′-UTR structure from the rest of the genome that might be necessary for efficient binding of viral RNA polymerase and cellular factors involved in transcription34 and may play a role in the natural transmission cycle of TBEV.44,45 A short poly(A) tract is genetically more stable compared with the virus having a long poly(A) tract.46
Previous studies reported that the variable region plays no role in viral replication and virulence for laboratory mice.43 However, recent studies revealed that partial deletions and poly(A) insertion in the variable region increases TBEV virulence in the mouse model.45,46 These data suggested that the variable region of the 3′-UTR might impact neurovirulence and function as a critical virulence factor.45,46
All TBFVs share a common folding pattern of secondary structures at the C3′-UTR position. RNA in this region is predicted to fold into a 3’ stem-loop and it contains conserved sequence elements. However, these structures are different from those observed in mosquito-borne flaviviruses.34 Indeed, some RNA sequences within the 3’-UTR clearly distinguish mosquito-borne from TBFVs.37,38 Modifications within the 3’-UTR of TBEV that affect the conserved structural motifs are known to attenuate the virus without altering their antigenic specificity. Modification of this region might form the basis for live-attenuated vaccines and/or for antiviral therapeutics.47,48
Short direct repeat sequences (20-70 nucleotides long) in the 3′-UTR were found to be conserved for each flavivirus group or subgroup.48 Four R1 repeats, two R2 repeats, and two R3 repeats, approximately 23, 26, and 70 nucleotides long, respectively, apparently arranged randomly, have been described in the 3′-UTR of the TBFVs.37,48 These short repeats apparently originated from at least 6 long repeat sequences (LRS) approximately 200 nucleotides in length, arranged in tandem. Four of these LRS are present in the 3′-UTR and 2 in the 3′ region of the ORF. Thus, it seems that evolution of the 3′-UTR and probably the ORF occurred through multiple duplications of LRS that form the basis for the development of the functionally important secondary RNA structures in the 3′-UTR. Subsequent formation of extended RNA domains evolved as promoters and enhancers of virus replication determined by the selective requirements of the vertebrate and invertebrate hosts.38,48
Flaviviruses, including TBFVs, are known to produce unique non-coding subgenomic flaviviral RNA (sfRNA), which is derived from the 3′-UTR. SfRNA results from incomplete degradation of viral RNA by the cellular 5’-3’ exoribonuclease XRN1.49 The exoribonuclease activity stops at the highly ordered RNA secondary structures at the beginning of the 3′-UTR. SfRNA is involved in modulating multiple cellular pathways; e.g., inhibiting antiviral activity of type I interferons (IFN) and RNAi pathways, facilitating viral pathogenicity.50
Proteins encoded by the virus
Structural proteins
C (Capsid) protein is a relatively small (11 kDa), basic, and highly positively charged protein with low sequence homology between different flaviviruses.39 Within the ORF that encodes the single polyprotein precursor of all structural and non-structural proteins, protein C is located at the amino-terminal end and is thus synthesized first during translation. The protein interacts with viral RNA genomes and represents a structural component of the nucleocapsid. Despite the low sequence homology among diverse flaviviruses, regions of hydrophobic and hydrophilic amino acids are conserved. The C-terminal hydrophobic domain (this domain is cleaved from mature C protein) is preceded by a hydrophilic region, and a central hydrophobic region. The N-terminus contains a hydrophilic region.31 The central hydrophobic region mediates membrane association of the protein and the charged residues that cluster at the hydrophilic N- and C-termini presumably mediate the interaction of the protein with viral RNA.39,51 In flavivirus infected cells, it was found that the mature C protein accumulates on the surface of endoplasmic reticulum (ER)-derived organelles named lipid droplets. The lipid droplets may play multiple roles during the viral life-cycle; i.e., they could sequester the flaviviral capsid protein early during infection and provide a scaffold for genome encapsidation.52
Figure 5
Click the image above to enlarge
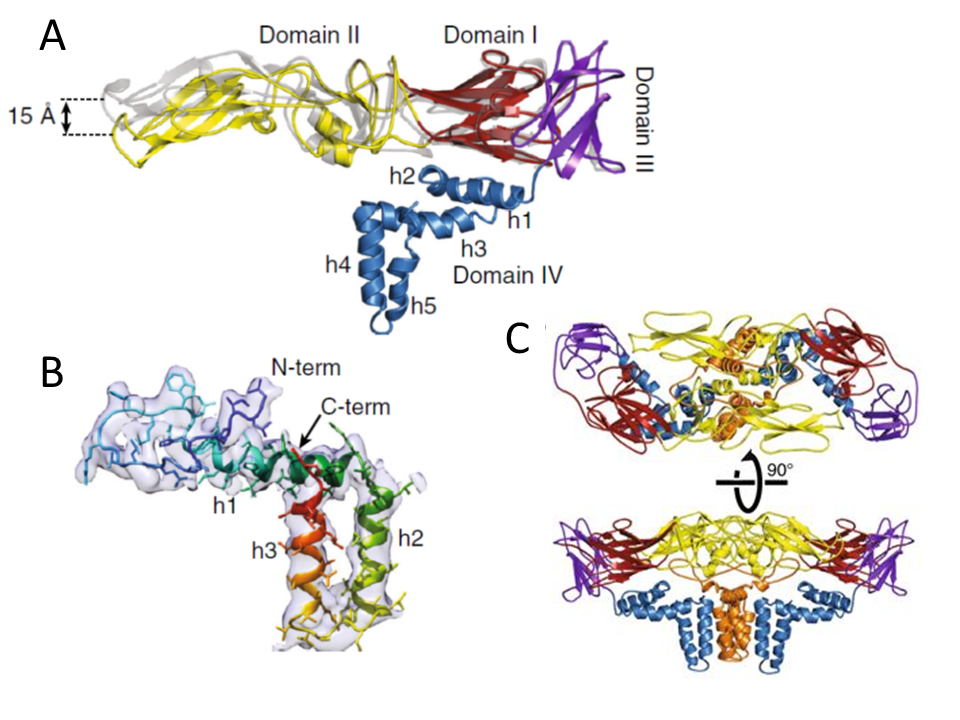
- Superposition of cryo-EM (colored) and X-ray (gray) E-protein structures. Domain I is colored in red, domain II in yellow, domain III in violet, and domain IV in blue.
- M-protein rainbow-colored from N-terminus in blue to C-terminus in red with electron density map shown as semi-transparent surface. The M-protein consists of an extended N-terminal loop followed by perimembrane (h1) and two transmembrane helices (h2 and h3).
- Heterotetramer of two E-proteins and two M-proteins. E-proteins are colored according to domains, and M-proteins are shown in orange.
Figures and figure legends are reproduced from Füzik et al. Nat Commun. 2018 Jan 30;9(1):436.
doi: 10.1038/s41467-018-02882-0 (https://www.nature.com/articles/s41467-018-02882-0) based on CC-BY 4.0 licence.
The introduction of various deletions into the TBEV genome that removed parts of the central hydrophobic domain of protein C revealed a remarkable structural and functional flexibility of this protein.53 TBEV mutants carrying deletions in C that extended from residue 28 up to residue 43 were viable in cell culture. The mutants produced substantial amounts of subviral particles lacking capsid, and the deletions impaired the assembly or stability of the virions.53 However, virus viability was affected when the deletions extended up to residue 48 or when the full hydrophobic domain was removed.53 Interestingly, these deletions led to spontaneous mutations in other regions of the C protein that generally increased the C protein hydrophobicity and restored infectivity of the virus.54
prM protein is a glycosylated precursor of the membrane protein M. The carboxyl terminus of C protein serves as an internal signal sequence element leading the structural protein prM into the membrane of the endoplasmic reticulum. The viral protease NS2B-NS3 cleaves this signal sequence, releasing the N-terminus of prM protein.53 The prM protein shows a chaperone-like activity during the envelope protein E folding.55 The N-terminus of the pr is mainly hydrophilic and, in TBEV, contains a single N-linked glycosylation site that appears to have an important role during virion assembly and release.31,39,56 Six cysteine residues, all disulphide-bridged, are highly conserved. The C-terminal region contains an ectodomain and 2 potential membrane-spanning domains.31 The cleavage of prM into pr and M occurs in the Golgi complex and is mediated by furin or a furin-like enzyme57,58 leading to a conversion from immature to mature fusogenic and fully infectious viral particles (Figure 3).57 The pr fragment is then secreted.39 A conserved region in the prM protein is a critical molecular determinant for the assembly and secretion of the virus.59 The M-protein consists of an N-terminal loop and three helices (Figure 5B). The first helix is situated as a perimembrane and the last two as trans-membranes; however, the M-protein is not exposed at the surface of the viral particle due to its small size and close association with the viral envelope membrane.118 Two M-proteins together with two E-proteins form a compact heterotetramer, which is the main building block of the virion, formed by head-to-tail dimerization of two E-M heterodimers (Figure 5C).118
The E protein contains the major viral antigens and is the main target for neutralizing antibodies (although antibodies directed against prM/M and NS1 also induce some protective immunity). Moreover, the E protein is responsible for specific binding to a cellular receptor and penetration of the virus into the host cell. It is also believed to be a main determinant of TBEV virulence.60 The three-dimensional structure of the E protein was studied at the resolution of 2.0 Å by X-ray crystallography61 (Figure 5). Comparison of the crystal structure of E protein and the structure of E protein in the virion observed by cryoelectron microscopy revealed root-mean-square deviations (RMSD) of 1.7 Å for the corresponding Cα atoms.118 The most important difference is in the positioning of domains I–III relative to each other. Whereas in the crystal structure the domains I, II, and III are arranged in a line, in the virion the tip of domain II is bent 15 Å towards the virus membrane (Figure 5A).118 Such a bending of the ectodomain in the virion prevents induction of premature membrane fusion mediated by the E protein.118 The structure of TBEV E protein was found to be highly similar to E1 glycoprotein from a distantly related virus, Semliki Forest virus (family Togaviridae). These proteins were defined as class II virus fusion proteins, distinct from previously characterized class I fusion proteins such as hemagglutinin of influenza virus.39
The protein forms 2 monomers anchored in the membrane by their distal parts at physiological pH. After virus uptake by receptor-mediated endocytosis into host cells, acidic pH in endosomes triggers irreversible changes in the E protein structure including its re-arrangement to trimeric forms. This leads to the initiation of the fusion process between the viral and endosomal membrane.62 Conserved histidines in the E protein function as molecular switches and, by their protonation at acidic pH, control the fusion process.63
Each E protein monomer is composed of 3 domains (I- III). Domain I is located in the central part of the protein. It is formed by 8 antiparallel beta sheets, contains the N-terminus of the protein, 2 disulphide bridges, and an N-glycosylation site. The function of E protein glycosylation was investigated using recombinant TBEV with or without the E protein N-linked glycan. The results suggested that glycosylation of the TBEV E protein is critical for the intracellular secretory process in mammalian cells but cleavage of the N-linked glycan after secretion did not affect virion infectivity in these cells. On the other hand, E protein glycosylation seems to play no significant role in virus reproduction in ticks.64
Domain II is formed of 2 long loops that extend out of domain I and form a finger-like structure. Domain II contains a number of beta sheets and 3 disulphide bridges.61,65 Part of the domain responsible for the fusion of viral envelope with the membrane of the endosome is called the fusion peptide; this peptide mediates insertion of the E protein into the endosomal membrane resulting in fusion of viral envelope with the membrane of the endosome.66 The initiation of fusion is crucially dependent on the protonation of 1 of the conserved histidines (His323), which works as a pH sensor at the interface between domains I and III of E, leading to the dissolution of domain interactions and to the exposure of the fusion peptide.63
Domain III has the typical fold of an immunoglobulin constant (IgC) molecule.65 It contains a beta barrel composed of 7 antiparallel beta sheets. The lateral part of domain III is believed to be responsible for binding to a specific cellular receptor.61
Amongst the most conserved parts of the E protein, there are 12 cysteine residues forming 6 disulphide bridges with conserved localization in common with all known flaviviruses.67
The E protein is also considered to be a major determinant of TBEV virulence. Amino acid substitutions in E protein often cause a decrease in neuroinvasiveness, although neurovirulence is usually not reduced.68 The highest number of attenuating mutations in the E protein was revealed in the domain that probably binds to specific cell receptors and participates in membrane fusion.62 A number of identified substitutions causing escape of the virus from the neutralizing effect of monoclonal antibodies,69 deficiency in the ability to agglutinate erythrocytes,70 and a change in virus growth properties in cell cultures, mice, or ticks,60,71-74 have been described.
Non-structural proteins
NS1 is a glycoprotein containing 2 or 3 potential glycosylation sites and 12 conserved cysteines forming disulphide bridges.75 It exists in dimeric forms localized freely in the cytoplasm or associated with membranes. Since the protein is highly hydrophilic and contains no transmembrane domains, its association with membranes remains poorly understood. Probably, dimerization creates a hydrophobic surface of the protein for its peripheral association with membranes.39,76 Alternatively, some species of the protein could be anchored into the membrane by glycosyl-phosphatidylinositol.39,77 The intra-cellular NS1 is central to viral RNA replication. The NS1 protein along with other non-structural proteins (see below) and viral RNA are targeted towards the luminal side of the endoplasmic reticulum, forming a replication complex (RC). Intracellular NS1 also interacts with various host proteins to assist viral replication, translation, and virion production; e.g., interaction of NS1 with 60S ribosomal subunits was described.78 Secretion of NS1 protein into the extracellular space appears particularly in the form of pentamers or hexamers and occasionally as decamers or dodecamers.79 This so-called ‘soluble antigen’, together with membrane-bound NS1 induces a protective immune response in the host.80 NS1 protein is also known to activate the Toll-like receptors (TLRs),81 and inhibit the complement system.82-83
NS2A is a small, hydrophobic protein, currently with no defined function. It is believed to play a role in forming the RC.39 A small membrane-associated protein, NS2B, serves as a crucial co-factor for protease activity of the NS3 protein. The central hydrophilic domain of the NS2B protein possibly interacts with the NS3 protein and it is flanked by hydrophobic regions probably anchored in the membrane.85 The central hydrophilic region of NS2B (40 amino acids that mediate the NS2B co-factor activity) is flanked by hydrophobic regions that mediate membrane association.39
NS3, the second largest viral protein, is an enzyme central to virus replication and polyprotein processing. Conserved regions impart functions as a serine protease, helicase, and RNA nucleoside triphosphatase.39 The protease activity is localized at the N-terminal domain of NS3, and this enzyme cleaves peptide bonds between NS2A-NS2B, NS2B-NS3, NS3-NS4A, and NS4B-NS5. As mentioned above, the protease activity occurs, in association with a 40-amino acid region of NS2B, resulting in the formation of a heterodimeric complex.39,86 It was found that mutations which were mapped in close proximity to the NS2B-NS3 protease active site may determine the neuro- or non-neuropathogenicity of TBEV.87 The C-terminal region of the NS3 protein has a helicase activity, utilizing the energy released from ATP to unwind RNA duplexes. Possible functions include elimination of complex secondary structures of viral RNA and/or resolving RNA duplexes formed during replication.39 The C-terminal region also has RNA triphosphatase and 5’RNA phosphatase activities.88 Due to the crucial role of NS3 protein in the virus replication process, this protein represents an excellent target for the development of specific antiviral inhibitors.86,89
NS4A and NS4B are small, hydrophobic proteins. NS4A is probably part of the replication complex.90 NS4B, a trans-membrane protein localized to the sites of replication and nucleus, partially blocks activation of STAT1 and IFN-stimulated response element (ISRE) promoters in cells stimulated with IFN.91 NS4A and, to a lesser extent, NS2A also block IFN signaling, and the cumulative effect of these 2 proteins together with NS4B results in robust IFN signaling inhibition.92
NS5 is the largest (100 kDa) and most highly conserved viral protein serving as a viral RNA-dependent RNA polymerase.93/sup>< Its C-terminus shares sequence homology with RNA-dependent RNA polymerases of other positive-stranded RNA viruses.39,94The N-terminal domain has a function as AdoMet-dependent methyltransferase involved in the mRNA capping process, transferring a methyl group from the cofactor S-adenosyl-l-methionine onto the N7 atom of the cap guanine and onto the 2’OH group of the ribose moiety of the first RNA nucleotide.86/sup>< The NS5 proteins form complexes with NS3 proteins, which results in stimulation of the NS3 RNA nucleoside triphosphatase activity.39,95
The NS5 protein is a promising target for specific antiviral inhibitors. Indeed, several nucleoside analogues targeting NS5 and causing premature termination of viral RNA synthesis were found to exhibit high inhibitory activity against TBEV.96,97
Apart from the main function as RNA-dependent RNA polymerase, the TBEV NS5 protein interferes with type I IFN JAK-STAT signaling.98,99
Figure 6
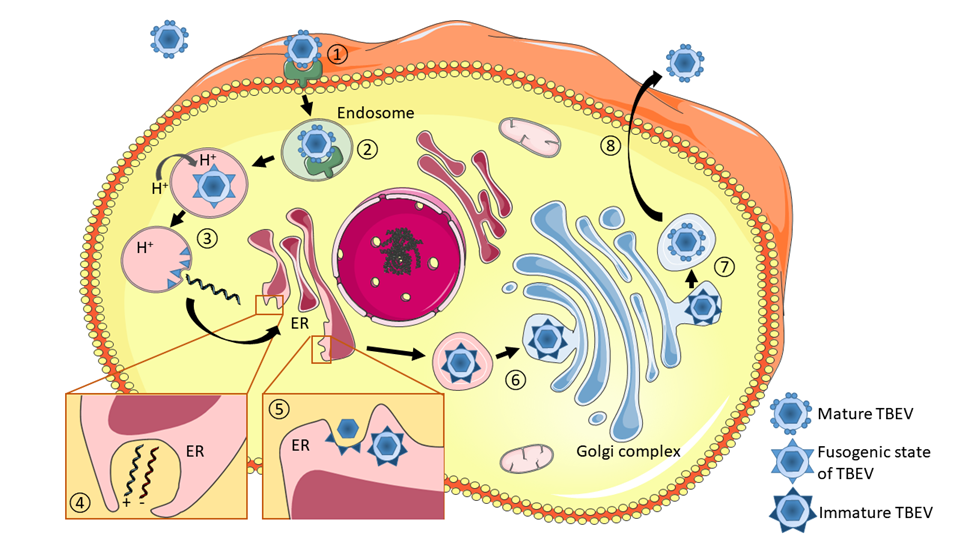
Click the image above to enlarge
Schematic illustration of the TBEV life cycle. (1) Infection begins with the binding of viral particles to specific cell-surface receptors, which have not yet been unequivocally identified. (2) Viral particles enter cells via endocytic pathway. (3) Low pH in the late endosome triggers conformational changes in the E proteins, leading to rearrangement of dimers to trimeric forms (fusogenic state) and the subsequent fusion of the viral envelope with endosomal membranes, which leads to virion uncoating. (4) Replication of the virus occurs through the synthesis of anti-sense (negative) RNA, which serves as the template for genome RNA production. Replication complexes are localized in membranous structures within the endoplasmic reticulum (ER). (5) Assembled nucleocapsids acquire lipid envelopes by budding into the ER lumen. (6) Immature particles pass through the Golgi complex. (7) Maturation takes place in the trans-Golgi network, involving the cleavage of prM and the reorganization of E proteins into fusion-competent homodimers, leading to a change from spiky immature to smooth mature particles. (8) Mature particles are transported in cytoplasmic vesicles and released into the extracellular space by exocytosis.
Reproduced from Ruzek et al., Antiviral Res. 2019 Jan 30. pii: S0166-3542(18)30447-9. doi: 10.1016/j.antiviral.2019.01.014. with permission from Elsevier.
Replication strategy
Infection of the host cell with TBEV begins with the binding of the virus to a cell receptor (Figure 6), which has not yet been unequivocally identified. Interaction of the viral particle with cellular receptors is mediated by viral E glycoprotein. Kopecký et al.100 identified 2 polypeptides of 35 and 18 kDa as putative vertebrate receptors for TBEV using a viroblot technique with anti-idiotypic monoclonal antibodies directed against antibodies that neutralize the infectivity of TBEV. However, the anti-idiotypic monoclonal antibodies did not bind effectively to tick cells, implying that different receptors are used by vertebrate and invertebrate cells for the binding of TBEV.100 It remains unclear whether TBEV uses single or multiple receptors on susceptible cells. Involvement of highly conserved glycosaminoglycans, such as heparan sulphate, during attachment and entry of flaviviruses has been suggested, but it seems likely that other host-cell receptor(s) can also mediate entry of TBEV into the host cells.101 Apparently, just the ability to use multiple receptors could be responsible for the very wide host range of flaviviruses, which replicate in arthropods and in a broad range of vertebrates.
Figure 7
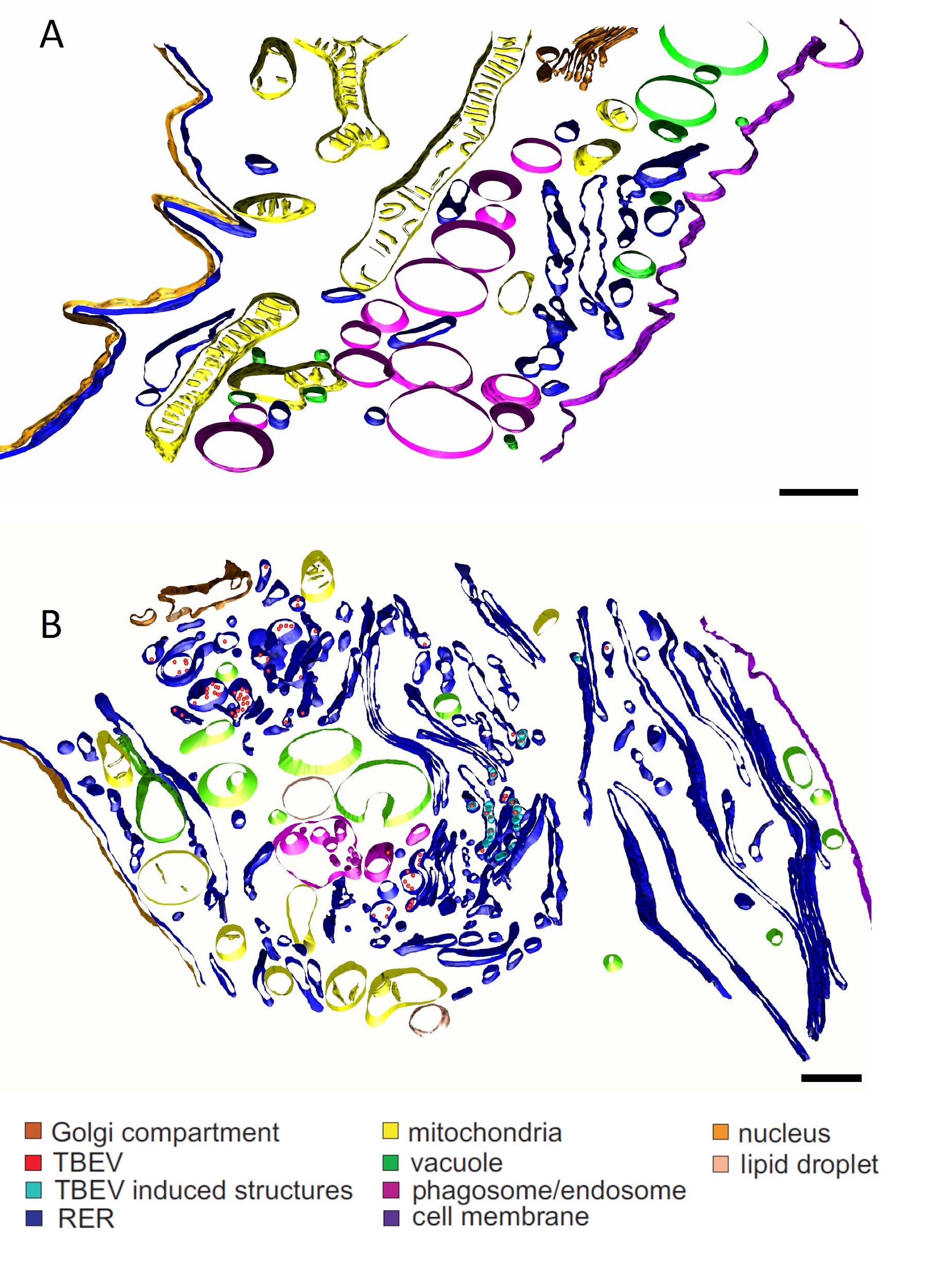
Click the image above to enlarge
Morphological changes in TBEV-infected mammalian cells. 3D models of mock-infected (A) and TBEV-infected human astrocytes (B). TBEV infection causes extensive morphological changes, including membrane reorganization of the endoplasmic reticulum; differences are evident in the Golgi complex, mitochondria, and phagosomes. (From Palus M, Bílý T, Elsterová J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. J Gen Virol 2014;95(Pt 11):2411-26, with permission).
In addition, in the presence of sub-neutralizing levels of specific immunoglobulins, the attachment and uptake by cells expressing Fc receptors might be enhanced, and this is called antibody-dependent enhancement.
After binding to the receptor, the virus is internalized into clathrin-coated vesicles by the process of endocytosis (see Chapter 2b for details). Acidification within the endosomal vesicle triggers conformational changes of the E proteins leading to rearrangement of the dimers to trimeric forms and subsequent fusion of the viral envelope with the membrane of the vesicle (Figure 6). The viral nucleocapsid is then released into the cytoplasm and viral RNA is uncoated. The exact mechanism of nucleocapsid uncoating remains unknown. The positive-sense viral RNA is the translational template, also functioning as a template for negative-sense RNA synthesis and formation of the double-stranded replicative intermediate.
The ratio of the newly synthesized positive-stranded RNA to negative-stranded RNA is at least 10 or 100 to 1, indicating that some regulatory mechanism must exist to produce higher numbers of positive-stranded RNA molecules.31 The biological explanation for this is the double function of the genomic positive-strand RNA: it is used as a template both for transcription of the negative strand and translation of the viral polyprotein, while the negative strand is only transcribed into the new positive strands.36
The single viral polyprotein is cleaved by viral and cellular proteases into individual viral proteins. The surface structural proteins prM and E (and also NS1) are translocated into the lumen of the ER and their amino termini are liberated through proteolytic cleavage by host signalase. The newly synthesized RNA is condensed by protein C into nucleocapsids on the cytoplasmic site of ER. Viral envelope is acquired by budding of the nucleocapsid into ER.102
TBEV replicates in the cytoplasm in close association with virus-induced intracellular membrane structures, also called replication compartments (Figure 6). These compartments provide an optimal microenvironment for viral RNA replication by limiting diffusion of viral/host proteins and viral RNA, thereby increasing the concentration of components required for RNA synthesis, and by providing a scaffold for anchoring the replication complex.103 These packets of vesicles have a diameter of about 80 nm and are formed as invaginations of the endoplasmic reticulum within a highly-organized network of interconnected membranes (Figure 6).103
The immature non-infectious virions containing proteins prM and E in heterodimeric association are transported to the Golgi complex, where the pr part of the prM molecule is cleaved, and the E protein is reorganized from trimers to form fusion-competent homodimers. These mature virions pass through the host secretory pathway and are finally released from the host cell by fusion of the transport vesicle membrane with the plasma membrane (Figure 6).102
TBEV infection is associated with dramatic morphological changes occurring in the infected cells (Figure 7). These include formation of smooth membrane structures, proliferation of endoplasmic reticulum, reorganization of the Golgi complex, and accumulation and convolution of membranes. Several cellular organelles are often damaged.104–107 The infection is commonly cytocidal; the infected cells often die by apoptosis or necrosis,104 but some vertebrate cell types survive the lytic crisis and become chronically infected.108
It was found that NS3 protein from Langat virus is able to activate cellular caspase-8 and induce apoptosis of the host cell.109 On the other hand, tick cells do not undergo major inhibition of host macromolecular synthesis caused by the infection. No dramatic cytopathic and ultrastructural changes are seen in the infected tick cells and persistent productive infection is established in these cells.107,110–113 However, both vertebrate and tick cells activate innate defense mechanisms against the infection.113
The TBEV maturation process in tick cells seems, however, to be different from that observed in vertebrate cells. In a cell line derived from the tick Rhipicephalus appendiculatus infected with TBEV, nucleocapsids are found in the cytoplasm and the envelope is acquired by budding on cytoplasmic membranes or into cellular vacuoles.114
Concluding remarks
The chapter summarized the major biological features of TBEV, focusing particularly on virus taxonomy, structure, genetics, and replication strategy in host cells. The past 2 decades have witnessed a tremendous progress in our understanding of the structural, biochemical, and molecular aspects of a variety of the processes involved in morphogenesis, genome replication, maturation, and genetic basis for virulence of flaviviruses, including TBEV.
This has been made possible by the recent advances in structural and biochemical techniques, and methods of molecular biology, mainly site-directed mutagenesis. However, several key questions related to TBEV molecular biology and individual steps in the TBEV life-cycle remain unresolved. Major gaps in our understanding of the TBEV replication strategy both in mammalian and tick cells still exist. For instance, the nature of the cellular receptor for virus entry into the host cell, mechanisms of viral genome release from nucleocapsid, packaging of viral RNA by the C protein, and virus maturation remain to be identified. Except for the E glycoprotein, no structural data for the other TBEV proteins are available, and indeed the complete functional role of some proteins remains obscure. The role of specific RNA secondary structures present in TBEV untranslated genomic regions in viral RNA replication, capping, and controlling the functions of non-structural proteins, such as NS3 or NS5, need to be established. These and other unresolved problems highlight the necessity for further research into the molecular, genetic, and structural properties of TBEV. Advances in our basic knowledge of TBEV biology should promote the development of more effective methods of controlling this important human pathogen.
Acknowledgments:
DR was supported by the Ministry of Health of the Czech Republic, grant No. 16-34238A. DR and KY were supported by the project Mobility Plus JSPS-18-08 funded by the Czech Academy of Sciences and Japanese Society for Promotion of Science. MEB was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, USA. We gratefully acknowledge the assistance of Dr John Pettersson (Zoonosis Science Center, Uppsala University, Sweden), who prepared and supplied Figure 1, Dr Tibor Füzik, who provided Figure 3D, and Dr Martin Palus, who provided Figure 4.
Contact:
ruzekd@paru.cas.cz
Citation:
Růžek D, Yoshii K, Bloom ME, Gould EA. Virology. Chapter 2a. In: Dobler G, Erber W, Bröker M, Schmitt HJ, eds. The TBE Book. 6th ed. Singapore: Global Health Press; 2023. doi:10.33442/26613980_2a-6
References
- Gould EA, Solomon T. Pathogenic flaviviruses. 2008;371(9611):500-9.
- Hollidge BS, González-Scarano F, Soldan SS. Arboviral encephalitides: transmission, emergence, and pathogenesis. J Neuroimmune Pharmacol. 2010;5:428-42.
- Wilson MR. Emerging viral infections. Curr Opin Neurol. 2013;26:301-6.
- Porterfield JS. 1980. Antigenic characteristics and classification of Togaviridae. In: The Togaviruses, pp. 13 46. Edited by Schlesinger RW. New York and London: Academic Press. 1980; pp. 13-46
- Blitvich BJ, Firth AE. Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. 2015;7:1927-59.
- Ergunay K, Tkachev S, Kozlova I, Růžek D. A review of methods for detecting tick-borne encephalitis virus infection in tick, animal, and human specimens. Vector Borne Zoonotic Dis. 2016;16:4-12.
- Lindquist L. Tick-borne encephalitis. Handb Clin Neurol. 2014;123:531-59.
- Yoshii K, Sunden Y, Yokozawa K, et al. A critical determinant of neurological disease associated with highly pathogenic tick-borne flavivirus in mice. J Virol. 2014;88:5406-20.
- Ecker M, Allison SL, Meixner T, Heinz FX. Sequence analysis and genetic classification of tick-borne encephalitis viruses from Europe and Asia. J Gen Virol. 1999;80:179-85.
- Zlobin VI, Demina TV, Mamaev LV, et al. [Analysis of genetic variability of strains of tick-borne encephalitis virus by primary structure of a fragment of the membrane protein E gene]. Vopr Virusol. 2001;46:12-6.
- Tkachev S, Demina TV, Dzhioev Yu P, et al. Genetic Studies of Tick-Borne Encephalitis Virus Strains from Western and Eastern Siberia. In: Flavivirus Encephalitis, Edited by Růžek D, pp. 235-54. InTech (open access). DOI: 10.5772/847
- Demina TV, Dzhioev YP, Verkhozina MM, et al. Genotyping and characterization of the geographical distribution of tick- borne encephalitis virus variants with a set of molecular probes. J Med Virol. 2010;82:965-76.
- Grard G, Moureau G, Charrel RN, et al. Genetic characterization of tick-borne flaviviruses: new insights into evolution, pathogenetic determinants and taxonomy. 2007;361:80-92.
- Hubálek Z, Pow I, Reid HW, Hussain MH. Antigenic similarity of central European encephalitis and louping-ill viruses. Acta Virol. 1995;39:251-6.
- Moureau G, Cook S, Lemey P, et al. New insights into flavivirus evolution, taxonomy and biogeographic history, extended by analysis of canonical and alternative coding sequences. PLoS One. 2015;10:e0117849.
- Zanotto PM, Gao GF, Gritsun T, et al. An arbovirus cline across the northern hemisphere. Virology. 1995;210:152-9.
- Zanotto PM, Gould EA, Gao GF, Harvey PH, Holmes EC. Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci U S A. 1996;93:548-53.
- Gould EA, de Lamballerie X, Zanotto PM, Holmes EC. Evolution, epidemiology, and dispersal of flaviviruses revealed by molecular phylogenies. Adv Virus Res. 2001;57:71-103.
- Pettersson JH, Fiz-Palacios O. Dating the origin of the genus Flavivirus in the light of Beringian biogeography. J Gen Virol. 2014;95(Pt 9):1969-82.
- Kuno G, Chang GJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus flavivirus. J Virol. 1998;72:73-83.
- Marin MS, Zanotto PM, Gritsun TS, Gould EA. Phylogeny of TYU, SRE, and CFA virus: different evolutionary rates in the genus Flavivirus. Virology. 1995;206:1133-9.
- Shiu SY, Ayres MD, Gould EA. Genomic sequence of the structural proteins of louping ill virus: comparative analysis with tick-borne encephalitis virus. 1991;180:411-5.
- Gould EA, de Lamballerie X, Zanotto PM, Holmes EC. Origins, evolution, and vector/host coadaptations within the genus Flavivirus. Adv Virus Res. 2003;59:277-314.
- Slávik I, Mrena E, Mayer V. Studies on Tick-borne encephalitis virus. II. Virus morphology and some data on virus structure. Acta Virol. 1970;14:8-16.
- Heinz FX, Stiasny K, Püschner-Auer G, et al. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. 1994;198:109-17.
- Zhang W, Chipman PR, Corver J, et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10:907-12.
- Allison SL, Tao YJ, O’Riordain G, et al. Two distinct size classes of immature and mature subviral particles from tick-borne encephalitis virus. J Virol. 2003;77:11357-66.
- Heinz FX, Allison SL, Stiasny K, et al. Recombinant and virion-derived soluble and particulate immunogens for vaccination against tick-borne encephalitis. 1995;13:1636-42.
- Heinz FX, Mandl CW. The molecular biology of tick-borne encephalitis virus. Review article. APMIS. 1993;101:735-45.
- Černý J, Selinger M, Palus M, et al. Expression of a second open reading frame present in the genome of tick-borne encephalitis virus strain Neudoerfl is not detectable in infected cells. Virus Genes. 2016;52:309-16.
- Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649-88.
- Tuplin A, Evans DJ, Buckley A, et al. Replication enhancer elements within the open reading frame of tick-borne encephalitis virus and their evolution within the Flavivirus genus. Nucleic Acids Res. 2011;39:7034-48.
- Thurner C, Witwer C, Hofacker IL, Stadler PF. Conserved RNA secondary structures in Flaviviridae genomes. J Gen Virol. 2004;85:1113-24.
- Gritsun TS, Venugopal K, Zanotto PM, et al. Complete sequence of two tick-borne flaviviruses isolated from Siberia and the UK: analysis and significance of the 5′ and 3′-UTRs. Virus Res. 1997;49:27-39.
- Proutski V, Gaunt MW, Gould EA, Holmes EC. Secondary structure of the 3′-untranslated region of yellow fever virus: implications for virulence, attenuation and vaccine development. J Gen Virol. 1997;78:1543-9.
- Proutski V, Gould EA, Holmes EC. Secondary structure of the 3′ untranslated region of flaviviruses: similarities and differences. Nucl Acid Res. 1997;25:1194-202.
- Gritsun TS, Gould EA. Origin and evolution of flavivirus 5’UTRs and panhandles: trans-terminal duplications? 2007;366:8-15.
- Wallner G, Mandl CW, Kunz C, Heinz FX. The flavivirus 3′- noncoding region: extensive size heterogeneity independent of evolutionary relationships among strains of tick-borne encephalitis virus. 1995;213:169-78.
- Lindenbach BD, Rice CM. Molecular biology of flaviviruses. Adv Virus Res. 2003;59:23-61.
- Mandl CW, Kunz C, Heinz FX. Presence of poly(A) in a flavivirus: significant differences between the 3′ noncoding regions of the genomic RNAs of tick-borne encephalitis virus strains. J Virol. 1991;65:4070-7.
- Formanová P, Černý J, Bolfíková BČ, et al. Full genome sequences and molecular characterization of tick-borne encephalitis virus strains isolated from human patients. Ticks Tick Borne Dis. 2015;6:38-46.
- Leonova GN, Belikov SI, Kondratov IG, Takashima I. Comprehensive assessment of the genetics and virulence of tick-borne encephalitis virus strains isolated from patients with inapparent and clinical forms of the infection in the Russian Far East. 2013;443:89-98.
- Mandl CW, Holzmann H, Meixner T, et al. Spontaneous and engineered deletions in the 3′ noncoding region of tick- borne encephalitis virus: construction of highly attenuated mutants of a flavivirus. J Virol. 1998;72:2132-40.
- Sakai M, Muto M, Hirano M, Kariwa H, Yoshii K. Virulence of tick-borne encephalitis virus is associated with intact conformational viral RNA structures in the variable region of the 3′-UTR. Virus Res. 2015;203:36-40.
- Asghar N, Lee YP, Nilsson E, et al. The role of the poly(A) tract in the replication and virulence of tick-borne encephalitis virus. Sci Rep. 2016;6:39265.
- Sakai M, Yoshii K, Sunden Y, et al. Variable region of the 3′ UTR is a critical virulence factor in the Far-Eastern subtype of tick-borne encephalitis virus in a mouse model. J Gen Virol. 2014;95:823-35.
- Gritsun TS, Gould EA. The 3′ untranslated region of tick-borne flaviviruses originated by the duplication of long repeat sequences within the open reading frame. Virology. 2006;354:217-23.
- Gritsun TS, Gould EA. Origin and evolution of 3’UTR of flaviviruses: long direct repeats as a basis for the formation of secondary structures and their significance for virus transmission. Adv Virus Res. 2007;69:203-48.
- Silva PA, Pereira CF, Dalebout TJ, Spaan WJ, Bredenbeek PJ. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J Virol. 2010;84:11395-406.
- Roby JA, Pijlman GP, Wilusz J, Khromykh AA. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses. 2014;6:404-27.
- Khromykh AA, Westaway EG. RNA binding properties of core protein of the flavivirus Kunjin. Arch Virol. 1996;141:685-99.
- Samsa MM, Mondotte JA, Iglesias NG, et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009;5:e1000632.
- Kofler RM, Heinz FX, Mandl CW. Capsid protein C of tick- borne encephalitis virus tolerates large internal deletions and is a favorable target for attenuation of virulence. J Virol. 2002;76:3534-43.
- Kofler RM, Leitner A, O’Riordain G, Heinz FX, Mandl CW. Spontaneous mutations restore the viability of tick-borne encephalitis virus mutants with large deletions in protein C. J Virol. 2003;77:443-51.
- Lorenz IC, Allison SL, Heinz FX, Helenius A. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J Virol. 2002;76:5480-91.
- Goto A, Yoshii K, Obara M, et al. Role of the N-linked glycans of the prM and E envelope proteins in tick-borne encephalitis virus particle secretion. 2005;23:3043- 52.
- Elshuber S, Allison SL, Heinz FX, Mandl CW. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick- borne encephalitis virus. J Gen Virol. 2003;84:183-91.
- Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475-81.
- Yoshii K, Igarashi M, Ichii O, et al. A conserved region in the prM protein is a critical determinant in the assembly of flavivirus particles. J Gen Virol. 2012;93:27-38.
- Gritsun TS, Holmes EC, Gould EA. Analysis of flavivirus envelope proteins reveals variable domains that reflect their antigenicity and may determine their pathogenesis. Virus Res. 1995;35:307-21.
- Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature. 1995;375:291-8.
- Holzmann H, Stiasny K, York H, et al. Tick-borne encephalitis virus envelope protein E-specific monoclonal antibodies for the study of low pH-induced conformational changes and immature virions. Arch Virol. 1995;140:213-21.
- Fritz R, Stiasny K, Heinz FX. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J Cell Biol. 2008;183:353-61.
- Yoshii K, Yanagihara N, Ishizuka M, Sakai M, Kariwa H. N- linked glycan in tick-borne encephalitis virus envelope protein affects viral secretion in mammalian cells, but not in tick cells. J Gen Virol. 2013;94:2249-58.
- Heinz FX. Molecular aspects of TBE virus research. 2003;21 Suppl 1:S3-S10.
- Heinz FX, Allison SL. Flavivirus structure and membrane fusion. Adv Virus Res. 2003;59:63-97.
- Nowak T, Wengler G. Analysis of disulphides present in the membrane proteins of the West Nile flavivirus. 1987;156:127-37.
- McMinn PC. The molecular basis of virulence of the encephalitogenic flaviviruses. J Gen Virol. 1997;78:2711-22.
- Holzmann H, Stiasny K, Ecker M, Kunz C, Heinz FX. Characterization of monoclonal antibody-escape mutants of tick-borne encephalitis virus with reduced neuroinvasiveness in mice. J Gen Virol. 1997;78:31-7.
- Khasnatinov MA, Ustanikova K, Frolova TV, et al. Non-hemagglutinating flaviviruses: molecular mechanisms for the emergence of new strains via adaptation to European ticks. PLoS One. 2009;4:e7295.
- Labuda M, Jiang WR, Kaluzova M, et al. Change in phenotype of tick-borne encephalitis virus following passage in Ixodes ricinus ticks and associated amino acid substitution in the envelope protein. Virus Res. 1994;31:305-15.
- Goto A, Hayasaka D, Yoshii K, et al. A BHK-21 cell culture-adapted tick-borne encephalitis virus mutant is attenuated for neuroinvasiveness. 2003;21:4043-51.
- Mandl CW, Allison SL, Holzmann H, Meixner T, Heinz FX. Attenuation of tick-borne encephalitis virus by structure-based site-specific mutagenesis of a putative flavivirus receptor binding site. J Virol. 2000;74:9601-9.
- Mandl CW, Kroschewski H, Allison SL, et al. Adaptation of tick-borne encephalitis virus to BHK-21 cells results in the formation of multiple heparan sulphate binding sites in the envelope protein and attenuation in vivo. J Virol. 2001;75:5627-37.
- Lee JM, Crooks AJ, Stephenson JR. The synthesis and maturation of a non-structural extracellular antigen from tick-borne encephalitis virus and its relationship to the intracellular NS1 protein. J Gen Virol. 1989;70:335-43.
- Muller DA, Young PR. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res. 2013;98:192-208.
- Jacobs MG, Robinson PJ, Bletchly C, Mackenzie JM, Young PR. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 2000;14:1603-10.
- Cervantes-Salazar M, Angel-Ambrocio AH, Soto-Acosta R, et al. Dengue virus NS1 protein interacts with the ribosomal protein RPL18: this interaction is required for viral translation and replication in Huh-7 cells. 2015;484:113-26.
- Gritsun TS, Liapustin VN, Shatalov AG, Lashkevich VA. Multiple forms of the NS1 protein as the main component of the nonvirion (“soluble”) antigen of the tick-borne encephalitis virus. Vopr Virusol. 1990;35:471-4.
- Gould EA, Buckley A, Barrett AD, Cammack N. Neutralizing (54K) and non-neutralizing (54K and 48K) monoclonal antibodies against structural and non-structural yellow fever virus proteins confer immunity in mice. J Gen Virol. 1986;67:591-5.
- Chen J, Ng MM, Chu JJ. Activation of TLR2 and TLR6 by dengue ns1 protein and its implications in the immunopathogenesis of dengue virus infection. PLoS Pathog. 2015;11:e1005053.
- Avirutnan P, Hauhart RE, Somnuke P, et al. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. 2011;187:424-33.
- Rastogi M, Sharma N, Singh SK. Flavivirus NS1: a multifaceted enigmatic viral protein. Virol J. 2016;13:131.
- Yamshchikov VF, Compans RW. Formation of the flavivirus envelope: role of the viral NS2B-NS3 protease. Virol J. 1995;69:1995-2003.
- Chambers TJ, Nestorowicz A, Amberg SM, Rice CM. Mutagenesis of the yellow fever virus NS2B protein: effects on proteolytic processing, NS2B-NS3 complex formation, and viral replication. Virol J. 1993;67:6797-807.
- Bollati M, Alvarez K, Assenberg R, et al. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res. 2010;87:125-48.
- Růzek D, Gritsun TS, Forrester NL, et al. Mutations in the NS2B and NS3 genes affect mouse neuroinvasiveness of a Western European field strain of tick-borne encephalitis virus. 2008;374:249-55.
- Wengler G, Wengler G. The NS 3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. 1993;197:265-73.
- Singh V, Somvanshi P. Structural modeling of the NS 3 helicase of Tick-borne encephalitis virus and their virtual screening of potent drugs using molecular docking. Interdiscip Sci. 2009;1:168-72.
- Uchil PD, Satchidanandam V. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J Biol Chem. 2003;278:24388-98.
- Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, et al. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol. 2005;79:8004-13.
- Muñoz-Jordan JL, Sánchez-Burgos GG, Laurent-Rolle M, García-Sastre A. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A. 2003;100:14333-8.
- Steffens S, Thiel HJ, Behrens SE. The RNA-dependent RNA polymerases of different members of the family Flaviviridae exhibit similar properties in vitro. J Gen Virol. 1999;80:2583- 90.
- Černý J, Černá Bolfíková B, Valdés JJ, Grubhoffer L, Růžek D. Evolution of tertiary structure of viral RNA dependent polymerases. PLoS One. 2014;9:e96070.
- Cui T, Sugrue RJ, Xu Q, et al. Recombinant dengue virus type 1 NS3 protein exhibits specific viral RNA binding and NTPase activity regulated by the NS5 protein. 1998;246:409 -17.
- Eyer L, Šmídková M, Nencka R, et al. Structure-activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res. 2016;133:119-29.
- Eyer L, Valdés JJ, Gil VA, et al. Nucleoside inhibitors of tick-borne encephalitis virus. Antimicrob Agents Chemother. 2015;59:5483-93.
- Best SM, Morris KL, Shannon JG, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol. 2005;79:12828-39.
- Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell Microbiol. 2008;10:696-712.
- Kopecký J, Grubhoffer L, Kovár V, Jindrák L, Vokurková D. A putative host cell receptor for tick-borne encephalitis virus identified by anti-idiotypic antibodies and virus affinoblotting. 1999;42:9-16.
- Kroschewski H, Allison SL, Heinz FX, Mandl CW. Role of heparan sulphate for attachment and entry of tick-borne encephalitis virus. Virology. 2003;308:92-100.
- Mandl CW. Steps of the tick-borne encephalitis virus replication cycle that affect neuropathogenesis. Virus Res. 2005;111:161-74.
- Miorin L, Romero-Brey I, Maiuri P, et al. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J Virol. 2013;87:6469- 81.
- Růzek D, Vancová M, Tesarová M, et al. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J Gen Virol. 2009;90:1649-58.
- Palus M, Bílý T, Elsterová J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. J Gen Virol. 2014;95:2411-26.
- Bílý T, Palus M, Eyer L, et al. Electron tomography analysis of tick-borne encephalitis virus infection in human neurons. Sci Rep. 2015;5:10745.
- Offerdahl DK, Dorward DW, Hansen BT, Bloom ME. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One. 2012;7:e47912.
- Mlera L, Offerdahl DK, Martens C, et al. Development of a model system for tick-borne flavivirus persistence in HEK 293T cells. 2015;6:e00614.
- Prikhod’ko GG, Prikhod’ko EA, Pletnev AG, Cohen JI. Langat flavivirus protease NS3 binds caspase-8 and induces apoptosis. J Virol. 2002;76:5701-10.
- Lawrie CH, Uzcátegui NY, Armesto M, Bell-Sakyi L, Gould EA. Susceptibility of mosquito and tick cell lines to infection with various flaviviruses. Med Vet Entomol. 2004;18:268-74.
- Růzek D, Bell-Sakyi L, Kopecký J, Grubhoffer L. Growth of tick-borne encephalitis virus (European subtype) in cell lines from vector and non-vector ticks. Virus Res. 2008;137:142-6.
- Bell-Sakyi L, Růzek D, Gould EA. Cell lines from the soft tick Ornithodoros moubata. Exp Appl Acarol. 2009;49:209-19.
- Weisheit S, Villar M, Tykalová H, et al. Ixodes scapularis and ixodes ricinus tick cell lines respond to infection with tick-borne encephalitis virus: transcriptomic and proteomic analysis. Parasit Vectors. 2015;8:599.
- Senigl F, Grubhoffer L, Kopecky J. Differences in maturation of tick-borne encephalitis virus in mammalian and tick cell line. 2006;49:239-48.
- Demina TV, Dzhioev YP, Verkhozina MM, Kozlova IV, Tkachev SE, Plyusnin A, Doroshchenko EK, Lisak OV, Zlobin VI. Genotyping and characterization of the geographical distribution of tick-borne encephalitis virus variants with a set of molecular probes. J Med Virol. 2010;82(6):965-76.
- Kozlova IV, Demina TV, Tkachev SE, Doroschenko EK, Lisak OV, Verkhozina MM, Karan LS, Dzhioev YP, Paramonov AI, Suntsova OV, Savinova YS, Chernoivanova OO, Ruzek D, Tikunova NV, Zlobin VI. Characteristics of the Baikal subtype of tick-borne encephalitis virus circulating in Eastern Siberia. Acta Biomedica Scientifica. 2018;3(4):53-60.
- Dai X, Shang G, Lu S, Yang J, Xu J. A new subtype of eastern tick-borne encephalitis virus discovered in Qinghai-Tibet Plateau, China. Emerg Microbes Infect. 2018;7(1):74.
- Füzik T, Formanová P, Růžek D, Yoshii K, Niedrig M, Plevka P. Structure of tick-borne encephalitis virus and its neutralization by a monoclonal antibody. Nat Commun. 2018;9(1):436.