
Chapter 4:
Pathogenesis of TBE with a
focus on molecular mechanisms
Andrea Kröger and Anna K Överby
Key Points
- In this chapter we describe the pathogenesis of tick-borne encephalitis virus (TBEV).
- To cause infection, TBEV needs to cross three different barriers; the physical, the innate and adaptive and the blood-brain barrier.
- The trigger of innate immune and adaptive immune responses, by TBEV is necessary to clear the infection.
- TBEV employs strategies to evade the innate immune response.
- Tools to study TBEV pathogenicity such as mouse knock-out models and reverse genetics are also discussed.
Overcoming the barriers of the host
The host has highly effective defense mechanisms against infections (Figure 1). The overwhelming majority of infections are normally blocked by physical barriers such as the skin, mucosal membranes, and stomach. However, this first barrier to TBEV is already overcome by the tick through direct injection of the virus into the skin of the host during blood feeding. This allows the first replication phase of the virus locally in the skin. The second barrier is the coordinated innate and adaptive immune response that reacts to infection. The innate immune response includes cell intrinsic defense mechanisms like apoptosis, autophagy, type I interferon (IFN) response, and innate cell-mediated responses, which are then followed by adaptive immune responses with a specific antibody response and stimulation of T cells that limit virus replication and which are involved in pathogenicity. If the virus overcomes the second barrier, it will spread to peripheral organs and cause viremia. The third barrier controls entry of the virus to the central nervous system (CNS), e.g., by the blood-brain barrier (BBB). If overcome, the virus will replicate in neurons and cause encephalitis and meningitis.
Initial infection, viral amplification, and spread
Very early during the tick feeding process TBEV particles are transmitted to the host via tick saliva. Tick saliva acts as a pharmacologically active compound which inhibits pain/itch response, contains anticoagulants, antiplatelet components, vasodilators, and immunomodulators,1,2 that enhance viral transmission and dissemination.3 Analysis of skin explants from tick-feeding sites reveals viral antigen in neutrophils, monocytes and skin-resident dendritic cells (DC).4 Although not proven, these cells are likely to serve as a vehicle for transport of the virus to draining lymph nodes. For other flaviviruses it was demonstrated that viral amplification in the lymph nodes results in viremia and spreads to peripheral tissues. The specific target cells for TBEV infection in peripheral tissues are not well defined, but are thought to be subsets of DCs, macrophages and possibly neutrophils.5
Neuroinvasion
TBEV is a neurotropic virus and neuropathogenesis depends on the ability of the virus to enter the CNS and propagate. General mechanisms of CNS invasion by neurotropic viruses are breakdown of the BBB, infection of cerebral endothelial cells, virus shedding from choroidal cells, axonal transport through olfactory receptor neurons, and retrograde transport along peripheral nerve axons, or transport by the “Trojan-horse” mechanisms by which virus is transported by infected cells. Although this process has been studied intensively for West Nile virus (WNV) infection,6 it is not known how TBEV reaches the CNS, but breakdown of the blood-brain barrier is unlikely because virus replication is detectable in the brain before BBB disruption.5,7
Figure 1: Barriers of TBEV infection

Click the image above to enlarge
Cellular responses to TBEV and implications for pathogenesis
Cell-intrinsic innate immunity
All cells have the capacity to react to various stresses, such as starvation, temperature extremes, irradiation, and infection. Cell-autonomous protective programs, which are inherent in all cells of the body are termed intrinsic cellular defenses.
Autophagy
Autophagy is a degradation pathway that occurs under stress conditions such as starvation, hypoxia, and infection. It starts with the sequestration of the area of the cytoplasm inside double-membrane vesicles called autophagosomes, which subsequently fuse with lysosomes to form autolysosomes or late endosomes.8 Dengue virus (DENV) infection promotes the formation of autophagy, which can enhance virus replication and protects cells against other stressors.9,10 Inhibition of dengue-induced autophagy by pharmacological inhibitors or deficiency of autophagy-related genes (ATG) reduces dengue replication. The importance of autophagy during TBEV replication was shown by stimulation of autophagy which results in significantly increased dose-dependent TBEV production, whereas the inhibition of autophagy showed a dose-dependent decrease of infectious virus.11
Apoptosis
Apoptosis is a process of programmed cell death in which cells activate intracellular death pathways.12 This mechanism occurs in a wide range of human viral infections, including infections of the CNS such as herpes simplex virus (HSV) and cytomegalovirus (CMV) encephalitis.13,14 In WNV infection of mice peak virus titers in the CNS are associated with the appearance of activated caspase 3 following infection, and apoptosis in neurons occurs in the same areas where viral antigen is present.15,16 In vitro, TBEV infection causes apoptosis in mouse and human neural cells.17,18 Although brain-infiltrating CD8+ T cells contribute to the fatal outcome during infection19 no significant increase of apoptotic cell death was detectable upon infection with Langat virus (LGTV) and TBEV in mice.5,20 These data are in line with human data, where no prominent signs of neuronal apoptosis were seen in post-mortem brain tissue from human patients.21
Figure 2: Viral evasion of IFN induction
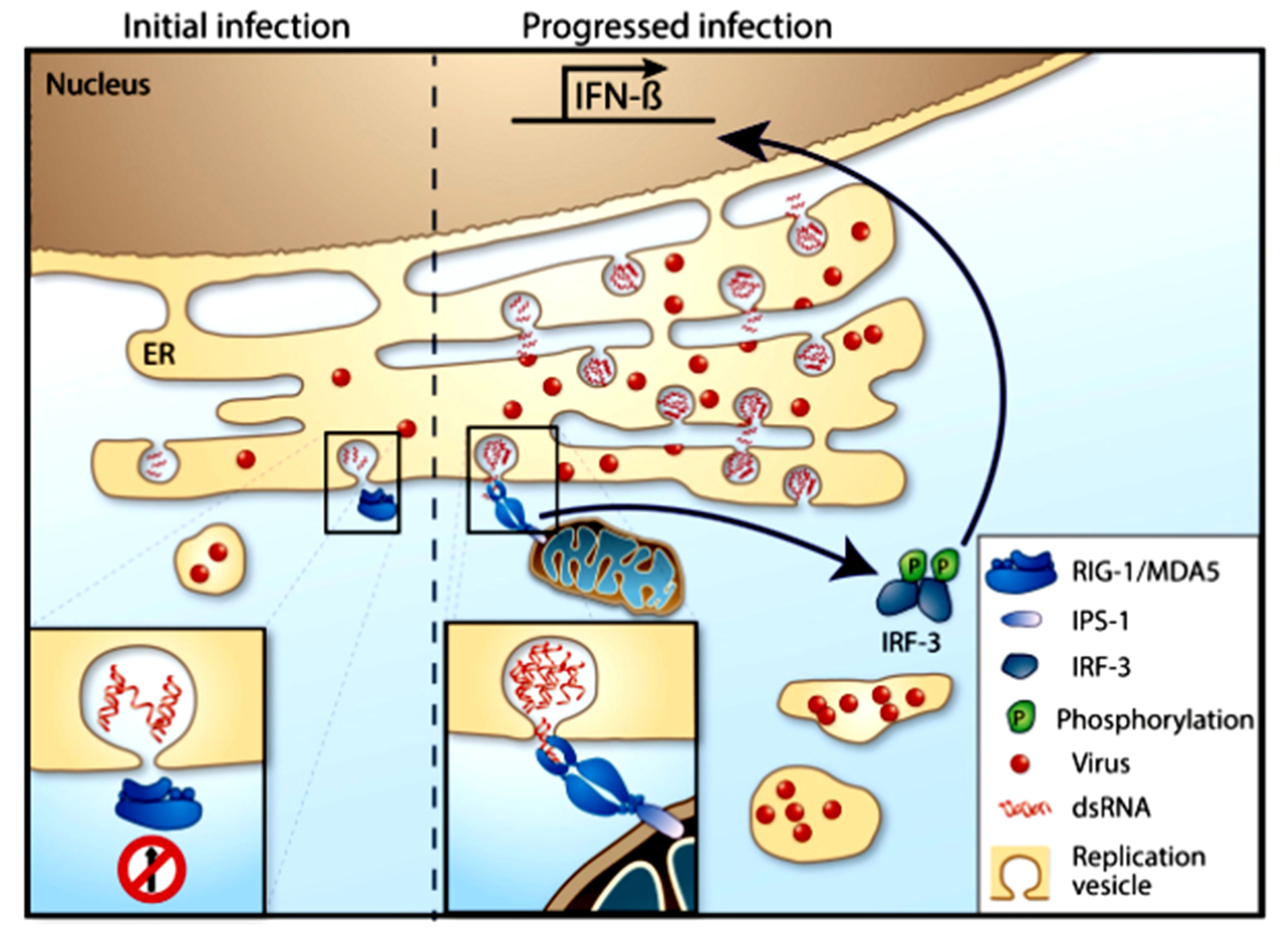
Click the image above to enlarge
Type I IFN response
The type I IFN system is the first line of defense against viral infection and an important part of the intrinsic innate immune response that controls virus dissemination and protects against serious disease. This response rapidly detects invading pathogens and upregulates inhibitory effector proteins and cytokines to ensure survival. The detection of pathogens is based on recognition of the non-self-pathogen-associated molecular pattern (PAMP) by specific host sensors, the pattern recognition receptors (PRR). This leads to a signaling cascade and the upregulation and secretion of IFN.22 IFN is a large family of cytokines where the IFNα and -β are type I IFNs and IFNγ is type II IFNs and these are the most studied. Type I IFNs binds to the IFNα receptor (IFNAR), which is expressed on nearly all cell types, in a paracrine and autocrine manner. The IFNAR is composed of a heterodimer of IFNAR1 and IFNAR2. After binding of IFN, the IFNAR activates the Janus kinases, Jak1 and Tyk2, which then phosphorylate the signal transducer and activator of transcription (STAT)-1 and STAT2 proteins, resulting in activation and translocation of the IFN-stimulated gene 3 (ISGF3) transcription factor complex into the nucleus. This ISGF3 induces hundreds of IFN stimulated genes (ISGs), that encode proteins with diverse biological function and some are potent antiviral proteins and part of the response against mammalian viruses.22
Figure 3: Viperin overexpression inhibits European TBEV growth by 4 orders of magnitude
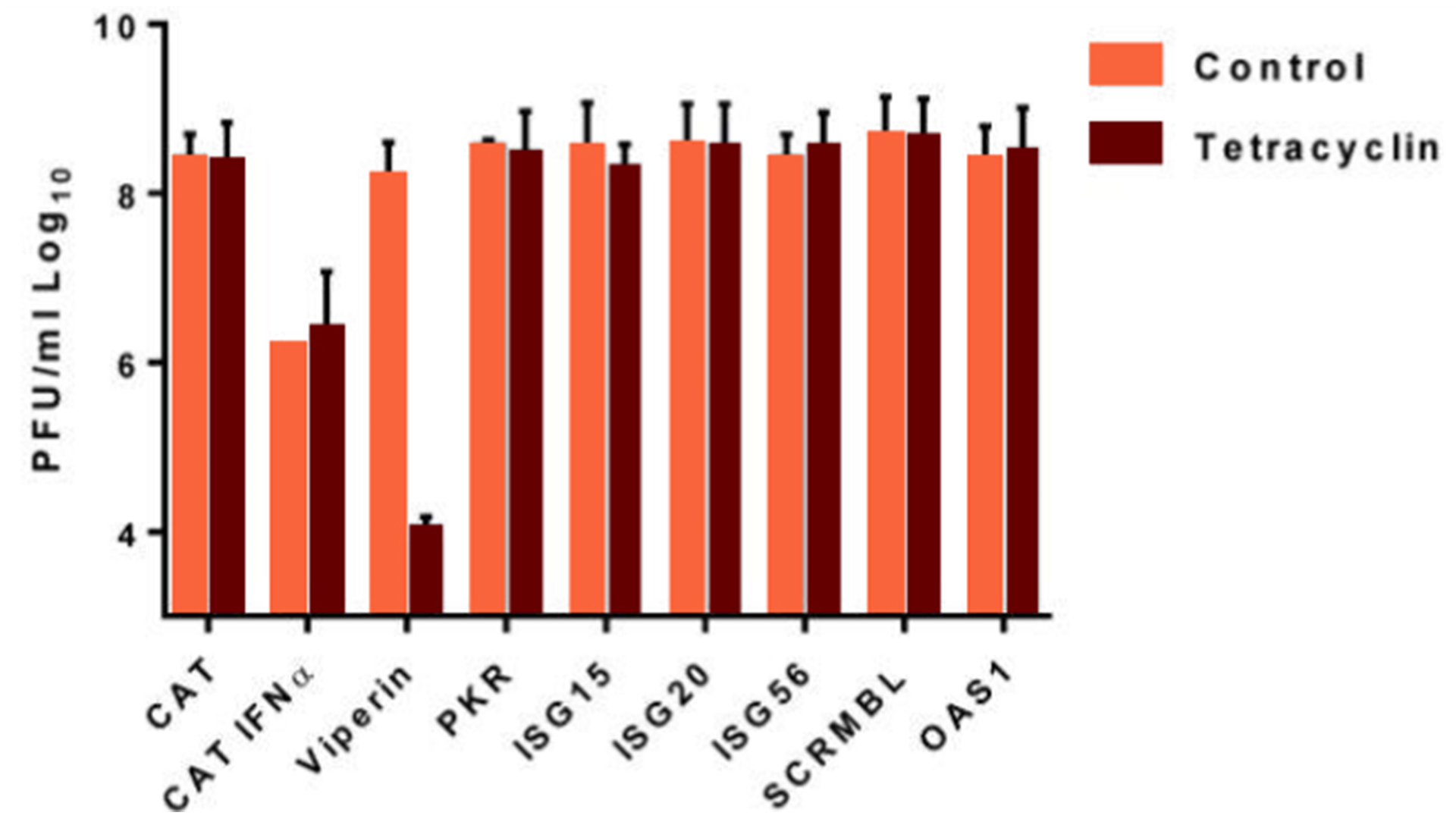
Click the image above to enlarge
Recognition of TBEV and induction of IFN
Rapid detection of the pathogen is crucial for mounting a protective response, and several different PRR families have been identified that recognize numerous ligands. The Toll-like receptors (TLRs) are located on the endosome or the plasma membrane, and the retinoic-acid-inducible gene I (RIG-I)-like receptors (RLRs) are in the cytosol. RNA viruses are most likely recognized by TLR3, TLR7, TLR8, or the RLRs (RIG-I and melanoma differentiation-associated gene 5, MDA5), which senses single-stranded RNA (ssRNA) or double-stranded RNA (dsRNA).23-25
For TBEV, it is not totally clear which PRRs are dominant. RIG-I, which recognizes short dsRNA and 5’ PPP, has been shown to be important for IFNβ induction in the U2OS (human osteosarcoma) cell line by siRNA depletion,26 however, the importance of MDA5 as contributing to sensing of TBEV cannot be ruled out as its involvement in sensing other flaviviruses has been demonstrated.27 Both RIG-I and MDA5 bind to the adaptor mitochondria-associated IFNβ promoter stimulator-1 (IPS-1, also called MAVS, VISA or CARDIF) via its caspase recruitment domain after binding to its RNA ligand. IPS-1 is important for IFNβ induction after TBEV infection in mouse embryonic fibroblasts (MEFs); in its absence, no IFNβ was detected.28 In addition, mice deficient in IPS-1 succumb to LGTV and TBEV infection earlier. These mice showed lower systemic levels of IFNα, resulting in higher viral titers in the periphery and leading to rapid invasion in the CNS.20 IPS-1 is also important in the local IFN response within the brain, reducing viral load and spread of LGTV,20,29,30 indicating an especially important role for RLR in the type I IFN response.
Upon IPS-1 activation, TNF Receptor Associated Factor 3 (TRAF3), TANK Binding Kinase 1 (TBK1) and Inhibitor-κB kinase ε (IKKε) are recruited, leading to phosphorylation and activation of the transcription factor IFN regulatory factor 3 (IRF3). Phosphorylated IRF3, dimerizes and translocates into the nucleus where it binds to the IFNβ gene promoter to initiate transcription and translation.31,32 IFNβ induction after TBEV infection has been shown to be highly dependent on IRF3 activation in the cells, and IRF3 has been shown to dimerize and translocate into the nucleus after TBEV infection.28
Very little is known about the importance of TLRs in TBEV infection, and only once the TLR7 has been investigated in the context of LGTV infection in vivo. This report demonstrates that mice deficient in TLR7 have higher viral load in the CNS and lower levels of pro-inflammatory cytokines. Primary neurons did not show a difference in infection rate, but TLR7 deficient neurons induced higher levels of IFNβ,33 indicating that TLR7 is more important for regulating neuroinflammation than type I IFNs.33
Since the type I IFN response is so important in controlling and restricting viral replication, most viruses have developed strategies to prevent upregulation of IFN by antagonizing the different steps in the IFN induction pathway. For example, dengue virus has been shown to reduce IFNβ levels by expressing the protease complex NS2B3,34 possibly by cleaving the adaptor STING.35 Dengue subtype 1/2/4 NS2A and NS4B and West Nile NS4B protein inhibited TBK1 phosphorylation and IFNβ induction.36 For TBEV, no specific IFN production antagonists have been identified among the different viral proteins.28 Instead, TBEV uses a passive escape mechanism that delays the induction of IFNβ by replicating inside replication vesicles or packets, thereby hiding its dsRNA from RIG-I and other PRRs.26,28,37,38 Later, during infection, the dsRNA leaks out from the replication vesicles, IRF3 is activated and translocates into the nucleus to transcribe IFNβ, which then is translated and secreted (Figure 2). Thus, the virus is produced and released from the cell before IFNβ can trigger an antiviral response in neighboring cells.28,38
Figure 4: Interferon (IFN) signaling and inhibition
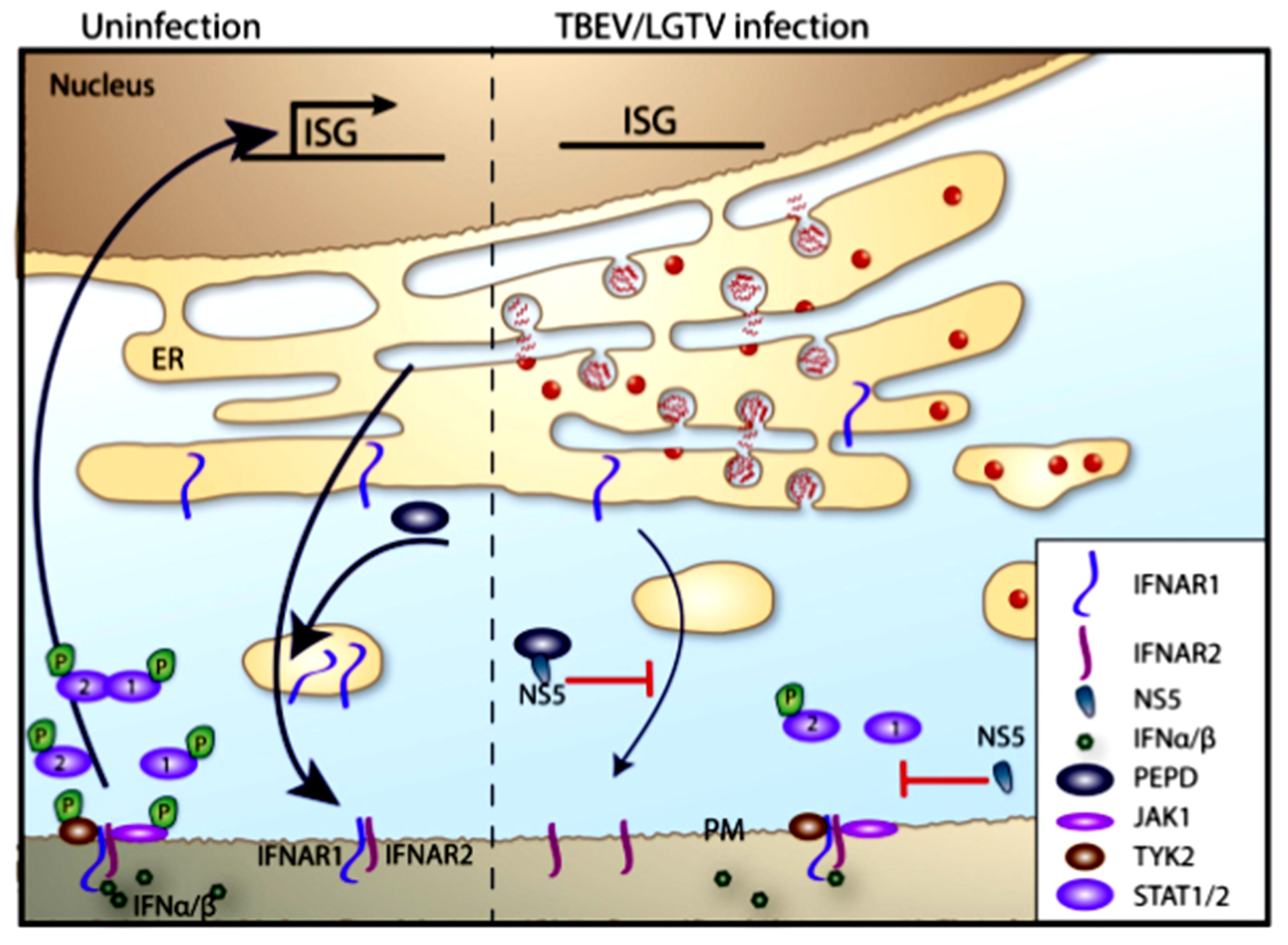
Click the image above to enlarge
Type I IFN signaling and response against TBEV
After infection and secretion of IFN, the IFN binds to its receptor the IFNAR1/2 which stimulates the upregulation of hundreds of ISGs that can limit the infection. The ISGs encode for PRR, adaptors and transcription factors to ensure a rapid response after infection. Cytokines and chemokines are also produced which activate and recruit immune cells to limit the infection, as well as antiviral proteins that can target viral replication directly in the cell.39 The IFNAR is therefore a key molecule in the type I IFN response. The importance of this molecule has been demonstrated for many viruses. For LGTV the type I IFN response determines tropism and can protect mice from lethal infection. In the absence of this response, the virus replicates uncontrollably in all organs, induces a rapid opening of the blood-brain barrier, and the mice succumb very quickly. This research has also shown that IFNAR is important in all cell types; hematopoietic, stroma, neuroectodermal and cells in the periphery.5
Most steps in the viral “life” cycle are targeted by 1 or several antiviral proteins encoded by the ISGs. Although several ISGs have been screened against TBEV (Figure 3), only 2 have been identified to be antivirally active so far; the rodent tripartite motif (TRIM) protein, TRIM79α, and viperin (virus inhibitory protein, endoplasmic reticulum-associated, IFN-inducible).40,41 The antiviral mechanism of TRIM79α is direct targeting of the viral polymerase, the non-structural protein 5 (NS5), an essential component of the replication complex, for lysosomal degradation. TRIM79α seems to be specific for TBEV and LGTV, because mosquito-borne flaviviruses; WNV and Japanese encephalitis virus (JEV), were shown not to be restricted by this protein.40 Viperin, on the other hand, is a highly conserved protein with broad spectrum antiviral activity, which has been shown to restrict a diverse range of viruses from different families. For the Flaviviridae family, viperin restricts hepatitis C, DENV, WNV and TBEV. However, the antiviral mechanism seems to depend on the specific virus. For TBEV, viperin selectively targets the positive stranded RNA synthesis. The intracellular location to the ER via viperin’s N-terminal amphipathic alpha helix is important as it coincides with viral replication. The antiviral activity is depending on the radical S-adenosyl methionine (SAM) domain and the proper iron-sulphur maturation of the protein.41,42 Recent studies have identified several viral and cellular interaction partners to viperin.42-47 Viperin is able to target TBEV in multiple ways mediating antiviral activity in a cell type-specific manner. Viperin interacts with several TBEV proteins; prM, E, NS2A, NS2B and NS3. The interaction between NS3 and viperin results in proteasome-dependent degradation of NS3.46 The stability of prM, E, NS2A and NS2B are affected by viperin, but only in the presence of NS3.46 Interestingly, although viperin does not directly interact with the TBEV C protein, viperin expression induces C particle formation and release from virus infected cells and disturbing the assembly process of TBEV.47 Viperin mediates this effect by interacting and sequestering the cellular protein Golgi brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1),47 which is involved in the vesicular trafficking of the secretory pathway48,49 and is a pro-viral factor for many different viruses.50-53 Thus, viperin may target other viruses via its interaction with GBF1. The in vivo importance of viperin during TBEV infection was recently shown in the viperin-/- mice.43 This study shows that specific regions of the brain rely differentially on the antiviral activity of viperin for protection against LGTV. Viperin is important in the olfactory bulb and cerebrum, while viral replication was unchanged in cerebellum and brain stem in the absence of viperin. This effect is due to the different neuronal subtypes, viperin expression is very important in cortical neurons but not at all in granular cell neurons isolated from the cerebellum.43 Although only 2 antiviral proteins have been identified so far, there are likely several others that are involved in the restriction and protection against TBEV and LGTV in vivo. One of the difficulties in identifying antiviral ISGs might be the redundancies seen between different proteins.
Even though different ISGs can potently restrict TBEV replication if induced before infection,40,41,54,55 IFN treatment after infection has limited effect in vitro.55 The reason for this is the expression of an IFN antagonist, NS5.55,56 The NS5 protein of LGTV interferes with the phosphorylation of Jak1 and Tyk2 in response to IFNβ, which leads to failure of STAT1/2 phosphorylation and subsequent ISG expression.55,56 Werme et al. showed that the interaction between Scribble and NS5 is important for plasma membrane targeting and IFN antagonist activity; however, the exact target of NS5 is unclear.56 In addition, NS5 was shown to block IFN signaling by selectively reducing the level of IFNAR1 expression on the cell surface. This reduction was dependent on NS5 binding to prolidase. Prolidase is needed for IFNAR1 intracellular trafficking, maturation, activation of IFNβ-stimulated gene induction, and IFN-I-dependent viral control (Figure 4).57 The relationship between NS5 function and virulence has not been observed for tick-borne flaviviruses, such as TBEV and the low virulence LGTV NS5; both exhibited the same degree of p-STAT inhibition. However, there are most likely other viral proteins that are important for pathogenicity and suppression of innate immune responses, as this has been shown for other flaviviruses. However, for TBEV these mechanisms have yet to be identified.
Complement
The complement system plays an essential role in the innate immune responses to many pathogens including flaviviruses. There is growing evidence that the complement system participates in the adaptive immune response. More than 30 proteins and protein fragments form a network of soluble and cell surface proteins that recognize and target pathogens. They orchestrate three distinct cascades: the classical pathway, alternative pathway, and lectin pathway. Each complement activation pathway is initiated by a distinct set of recognition molecules and converges at the cleavage of C3 to C3a and C3b. Beyond its lytic capacity, complement protects against viral infections by priming adaptive B and T cell responses, triggering leukocyte chemotaxis through the release of anaphylatoxins (C3a and C5a), and opsonizing viruses for phagocytosis and destruction by macrophages.58,59
Stimulation of all complement activation pathways contributes to protection against flaviviruses. For WNV infections enhanced susceptibility was shown for mice deficient in various components of the complement system. Less is known about the complement activation during TBEV infection. Antibody-dependent, complement-mediated cytolysis of infected cells is considered a possible mechanism of protection by NS1 antibodies, since NS1 is expressed on the cell surface.60 In response to these protective functions, many viral pathogens have evolved evasion strategies to limit recognition by and activation of the complement cascade. NS1 proteins of different flaviviruses limit complement activation by forming complexes with C1s and C4 to promote cleavage of C4 to C4b. Another mechanism shows direct interaction of NS1 with C4b binding proteins which leads to reduced C4 activity.58 Although these inhibitory mechanisms are functional in various flavivirus strains, less is known about the role of NS1 protein from TBEV.
Innate and adaptive immune interface
Natural killer (NK) cells
Natural killer (NK) cells are large granular lymphocytes that play an important role in the control of viral infections. NK cells limit viral replication by killing infected cells during early stages of infection. The antiviral response of NK cells includes direct killing of virus–infected cells, which is primarily mediated by perforin and granzyme, as well as the production of several proinflammatory cytokines, including IFN-γ and tumor necrosis factor (TNF).61 These molecules are components of the innate immune response as they are activated by type I IFNs, but they also play a critical role in immunoregulation during the development of adaptive immunity, thereby bridging innate and adaptive immune responses. Their important role in the host defense against viruses is supported by the finding that humans with complete or partial impairment of NK cell numbers and functions have increased susceptibilities to viral infections, including HSV, varicella zoster virus, CMV, and human papilloma virus.62
NK cells have been studied in various flavivirus infections including DENV, WNV, JEV and yellow fever virus (YFV). NK cells have been suggested to affect disease severity and outcome, as well as to contribute to viral control, even though the underlying mechanisms remain unknown.63-65 The role of NK cells in immunopathology of TBEV infection is largely unknown. Langat or TBEV infection in mice leads to a temporary activation of NK cells during the early phase of infection, followed by suppression,66 which in later phases of infection was not associated with increased viral replication in splenocytes. Ex vivo infection of whole-blood cells showed activation of NK cells only with low pathogenic TBEV strains while highly pathogenic TBEV inhibits NK cell activation. Decreased expression of perforin and granzyme B was detected in activated CD56dim NK cells of TBEV-infected patients during hospitalization, indicating that cytotoxic granules were released early in NK cell activation and symptom onset, thereby possibly contributing to pathogenesis of infection.67 Given these ostensibly conflicting results, more investigation is needed to determine the functional role of NK cells in limiting viral replication and in the pathology associated with TBEV infection in different hosts.
Antigen-presenting cells
Effective host defense against infection requires innate and adaptive immune responses working together to mediate clearance of invading pathogens. Dendritic cells (DCs) bridge these 2 arms of immunity. In peripheral tissues, immature DCs recognize RNA virus infection, migrate to local lymphoid tissues, and undergo a process of maturation that involves cytokine production and antigen presentation to activate naïve T cells and shape adaptive immunity.68 Many flaviviruses including DENV,69 WNV,70 and JEV71 infect DCs resulting in impaired DC maturation and T cell priming/proliferation and promoting viral pathogenesis. DCs also represent early targets of TBEV infection following the bite from an infected tick,4 providing the virus with opportunities to manipulate DC functions as a means of evading host immunity. LGTV infection impairs DC maturation by suppression of costimulatory molecules and inhibition of IL-12 production. This immature DC phenotype was associated with an impaired functional capacity to induce T cell proliferation.72 However, how this is involved in viral pathogenesis is unknown.
Adaptive Immune response to TBEV
Humoral immunity
Humoral immunity is an important component of the immune response. As with other flaviviruses, a functional humoral immune response is critically important in controlling infections.73 Passive transfer of monoclonal or polyclonal TBEV-specific antibodies protects mice in vivo and protection correlates with in vitro neutralization.74-77 No infectious virus could be detected in the blood or brain of passively protected mice subsequent to TBEV challenge. However, antibodies protect not only by neutralization; therefore, because limited virus replication does occur, this indicates that mechanisms of protection from disease exist other than sterilizing immunity.78
Cellular Immunity
In addition to effective humoral immunity, the activation of cellular immunity is usually required for clearance of established infection. Distinct T cell subsets play a key role in the induction of protective immune response against TBEV infections. CD4+ T cells are essential in priming the TBEV-specific antibody response and sustaining the CD8+ T cell response. However, results from studies in mice lacking B cells or CD4+ T cells during TBEV infections are missing. Nonetheless, mice lacking type I IFN signaling develop a normal antibody response during LGTV infection but are not protected from severe infection.5,20
Cytotoxic T lymphocytes (CTL) recognize viral peptides presented on major histocompatibility complex (MHC) class I molecules and eliminate cells producing abnormal or foreign proteins, specifically virus infected cells. CD8+ CTLs control viral replication via distinct mechanisms: non-cytolytically by secretion of IFN-γ or TNFα or cytolytically by cytotoxic proteins like granzyme B and perforin.79 Long-term immune surveillance effector cells react more quickly against the same virus after a primary infection.
The effects of TBEV infection on T cells are less studied. Ex vivo infection of human blood cells leads to an activated phenotype of T cells with low-pathogenic TBEV, whereas the highly pathogenic TBEV suppresses T-cell activation.80 It is unclear whether T cells are directly infected by TBEV, but no infection of T cells was detectable in highly susceptible IFNAR mice infected by Langat virus,5 which makes direct infection of T cells unlikely.
Studies in humans showed that CD8 T cells responded strongly to acute TBEV infection and passed through an effector phase, prior to gradual differentiation into memory cells, indicating that TBEV infection induces a robust CD8 T cell response.81 Comparable studies in mice revealed that the number and activation of T cells in the CNS have no impact in the outcome of infection; both dying and recovering mice showed no difference in number and activation status of T cells upon TBEV infection. However, differences were seen in the specific T cell clones accumulating in the brain.82
Besides their role in antiviral response, CD8+ T cells are also believed to contribute to CNS pathogenesis. In brain autopsy samples from TBEV-diagnosed individuals, inverse topographical correlation of inflammation and TBEV-infected areas has been reported.83 Inflammatory infiltrates are predominantly composed of T cells and macrophages/microglia. In regions with less infiltration CTL are closely associated with TBEV-infected neurons. These findings suggest that immunologic mechanisms can contribute to nerve cell destruction in human disease. In immune deficient SCID mice or mice lacking CD8 T cells an increased survival upon TBEV infection was shown. Adaptive transfer of CD8+ T cells in SCID mice decreases median survival time. Although these data suggest a contribution of CD8 T cells in pathogenesis, surprisingly, this effect is independent of viral replication in the periphery and the CNS. The pathogenicity of virus strains also seems to influence the effect of CD8 T cells on the outcome of infection. Whereas CD8+ T-cell-deficient SCID mice succumb later from infection with high pathogenic TBEV strains, a survival advantage was shown upon infection with low pathogenic strains.19
Although viral infection with LGTV leads to an accumulation of CD4+ and CD8+ T cells in the CNS, no increased numbers of apoptotic cells were detectable.5,20
Other data suggest that T cells within the CNS promote survival. In CCR5-deficient mice, an increase of viral replication in the CNS and decreased survival is due to the lack of lymphocyte migration to the CNS. Adaptive transfer of LGTV-specific T cells improved survival outcome. However, whether the protective effect is only mediated by T cells or by the decrease of inflammatory neutrophils in the presence of T cells is not clear.84 Because TBEV-infected mice also died of encephalitis in the absence of T cells, other cells such as neutrophils could contribute to pathogenic effects of TBEV infection. Further investigation is needed to better understand the processes that control the protective rather than pathogenic CD8+ T cell response during TBEV infection.
Tools to study pathogenesis
Mouse models
Laboratory mice are a useful tool to investigate human diseases, as mice are phylogenetically related to humans and show a striking genomic homology. This is especially true with knockout mice, in which an existing gene is inactivated. Laboratory mice are used to better understand how a similar gene in humans may cause or contribute to disease. The mouse as a model system for studying pathogenesis of TBEV has an advantage compared with other flaviviruses, because mice are susceptible to natural TBEV isolates, and develop encephalitis, whereas other flaviviruses require mouse adaptation to cause disease.85 Animal models of TBEV infections have provided insights into the pathogenesis of TBE in humans. In particular, TBEV and LGTV infections of mice enable the identification of host and viral genetic factors that contribute to the outcome of infection, as shown through the studies described elsewhere and in this chapter.
Recently we used C57BL/6 mice to characterize TBEV pathogenesis. Two different strains showing different symptoms are investigated. Namely HB171/11, isolated from questing adult ticks from a natural focus in south Germany86 and Torö-2003, rescued from a cDNA infectious clone generated from RNA extracts of nymphs collected in the island of Torö, Sweden.87 Both strains showed highly different symptoms in humans, as HB171/11 leads to mild gastrointestinal and constitutional symptoms without affecting the nervous system. TBE cases in the region of Torö showed relatively mild neurologic disease and few cases of hospitalization. The infection of mice reflects the different course of infection in humans, we observed lower pathogenicity of HB171/11 in comparison to Torö-2003 infections. Torö-2003 replicates faster in the periphery and enters the brain very early during infection. In addition, neurovirulence was lower in HB171/11-infected mice. The mechanism of virulence and neuropathology is still under investigation, although differences in cytokine induction and viral replication in target cells could be involved. In summary, mouse models could be a good tool to contribute to our understanding of pathogenesis of TBEV infection.88
Reverse genetics systems
Reverse genetics of viruses is the generation and manipulation of viral genomes to investigate the direct effects of changes on virus biology and pathogenesis. For flaviviruses, the first reverse genetic system was developed in 1989 for YFV.89 Since the genome of flaviviruses is positive stranded, they are infectious if introduced into susceptible cells.90 There are several different approaches to generate infectious virus. One important step is the generation of a complimentary DNA (cDNA) to the RNA genome. The cDNA is often cloned into a plasmid under a specific promoter, which enables the in vitro transcription of viral RNA. This DNA clone enables the introduction of mutations into the genome, and subsequent analysis of the resulting phenotype. Reverse genetics have been used to study virulence, replication, host range, vaccines, and functions of the coding and non-coding regions. However, these clones are laborious and difficult to generate due to instability and toxicity of some viral sequences in bacteria.91
For TBEV 2 separate approaches were used in the beginning; plasmid-based infectious clones92 and the PCR-based methods for constructing recombinant virus.93,94 Both rely on in vitro transcription and transfection of RNA. The most recent technique for generating TBEV clones is the infectious-subgenomic-amplicon (ISA) method. Three PCR amplicons are produced that have a CMV promoter at the 5′ non-coding region (NCR) and 70-100 bp overlapping regions; the hepatitis delta ribozyme is followed by the simian virus 40 polyadenylation signal. The amplicons are mixed and introduced into the cells where they recombine and produce infectious virus.95
Infectious clone systems have been very useful in studying determinants of replication and biological characteristics as well as to identify pathogenicity factors of TBEV. Two advantages of this approach are that the genome is defined and can be manipulated. In contrast, natural viral isolates of positive-stranded RNA viruses are present as a population of different viral types also called quasispecies. This is due to the error-prone RNA-dependent RNA polymerase. In addition, manipulating natural viral isolates with specific mutagenesis-inducing drugs is a very nonspecific approach.
With this technique, several determinates of pathogenicity have been identified. Specifically, the envelope protein responsible for receptor-mediated entry,96 the function of the membrane protein in virus budding,97 and the importance of different regions in the 3’NCR. Neurovirulence in mice was shown to be dependent on specific amino acid residues in the upper lateral surface of domain III in the envelope (E) protein of TBEV (residues E308, E310 and E311), possibly due to disruption of the receptor binding.96 The residues S267L, K315E, N389D in LGTV E protein and K46E in the NS3 protein, were shown to be crucial for neuroinvasiveness in immunodeficient mice.98 The 5’ and the 3’ NCR contain complementary sequences that help genomic cyclization to form panhandle structures. The NCRs have several conserved structural stem loops that are important for replication, translation initiation and packaging.99,100 At the beginning of the flavivirus 3’ NCR, a secondary structure forms a pseudoknot that protects the terminal 300- to 500-bases from exoribonuclease XRN1 degradation, generating a subgenomic flavivirus RNA (sfRNA) 101-103. The sfRNA has been shown to be critical for WNV induced cytopathic effects104 and pathogenicity in mice,104 and is involved in viral subversion of type I IFN response by a yet unknown mechanism.105 The TBEV sfRNA has been shown to specifically interfere with the RNAi system of ticks.106 The 3’ NCR of TBEV can be divided into a highly conserved core element and a variable region that is both heterogenic in length and sequence.107 Several European TBEV strains contain an internal poly(A) tract in the variable region of the 3’ NCR, which was considered dispensable for replication and virulence in mice.108,109 However, studies recently showed that the variable region and the poly(A) tract can modulate virulence of the Far Eastern TBEV.110,111 We have also detected different lengths of the poly(A) tract in a blood-feeding tick indicating that the poly(A) might be important for the switch between invertebrate to vertebrate.112 To investigate this further a long-poly(A) Torö-38A and a TBEV Torö with a short-poly(A) were cloned and rescued. We were able to show that the viruses with long-poly(A) were attenuated in cell culture but more virulent in mice compared with the short-poly(A), and the genome with short-poly(A) was much more stable compared with the long version, which developed a high quasispecies diversity.87
Conclusion
Important advances in the identification of molecular and cellular mechanisms of TBEV-induced pathogenesis have been made in recent years. Nevertheless, many questions remain unresolved. The interaction of the virus with the innate and adaptive immunity is not fully understood. Additional questions include: which genes act antivirally to inhibit virus replication in the periphery and in the CNS? Are there cell- and tissue-specific differences? What is the effect of cells of the innate and adaptive immune system in antiviral defense and which factors influence neuroinvasion and neuropathogenesis? And, last but not least, how can CNS infections be prevented or treated?
Contact:
andrea.kroeger@med.ovgu.de
Citation:
Kröger A, Överby AK. Pathogenesis of TBE with a focus on molecular mechanisms. Chapter 4. In: Dobler G, Erber W, Bröker M, Schmitt HJ, eds. The TBE Book. 6th ed. Singapore: Global Health Press; 2023. doi: 10.33442/26613980_4-6
References
- Wikel S. Ticks and tick-borne pathogens at the cutaneous interface: host defenses, tick countermeasures, and a suitable environment for pathogen establishment. Front Microbiol. 2013;4:337.
- Kazimirova M, Stibraniova I. Tick salivary compounds: their role in modulation of host defences and pathogen transmission. Front Cell Infect Microbiol. 2013;3:43.
- Labuda M, Jones LD, Williams T, Nuttall PA. Enhancement of tick-borne encephalitis virus transmission by tick salivary gland extracts. Med Vet Entomol. 1993;7(2):193-196.
- Labuda M, Austyn JM, Zuffova E, et al. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology. 1996;219(2):357-366.
- Weber E, Finsterbusch K, Lindquist R, et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. Journal of virology. 2014;88(21):12202-12212.
- Suthar MS, Diamond MS, Gale M, Jr. West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11(2):115-128.
- Ruzek D, Salat J, Singh SK, Kopecky J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One. 2011;6(5):e20472.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728-741.
- Lee YR, Lei HY, Liu MT, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374(2):240-248.
- McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. The Journal of biological chemistry. 2011;286(25):22147-22159.
- Bily T, Palus M, Eyer L, Elsterova J, Vancova M, Ruzek D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci Rep. 2015;5:10745.
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-257.
- Perkins D, Gyure KA, Pereira EF, Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J Neurovirol. 2003;9(1):101-111.
- DeBiasi RL, Kleinschmidt-DeMasters BK, Richardson-Burns S, Tyler KL. Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. The Journal of infectious diseases. 2002;186(11):1547-1557.
- Samuel MA, Morrey JD, Diamond MS. Caspase 3-dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. Journal of virology. 2007;81(6):2614-2623.
- Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg Infect Dis. 2001;7(4):714-721.
- Isaeva MP, Leonova GN, Kozhemiako VB, Borisevich VG, Maistrovskaia OS, Rasskazov VA. [Apoptosis as a mechanism for the cytopathic action of tick-borne encephalitis virus]. Vopr Virusol. 1998;43(4):182-186.
- Ruzek D, Vancova M, Tesarova M, Ahantarig A, Kopecky J, Grubhoffer L. Morphological changes in human neural cells following tick-borne encephalitis virus infection. The Journal of general virology. 2009;90(Pt 7):1649-1658.
- Ruzek D, Salat J, Palus M, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. 2009;384(1):1-6.
- Kurhade C, Zegenhagen L, Weber E, et al. Type I Interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by IPS-1. J Neuroinflammation. 2016;13(1):22.
- Gelpi E, Preusser M, Laggner U, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. 2006;12(4):322-327.
- Weber F, Kochs G, Haller O. Inverse interference: how viruses fight the interferon system. Viral Immunol. 2004;17(4):498-515.
- Nazmi A, Dutta K, Hazra B, Basu A. Role of pattern recognition receptors in flavivirus infections. Virus research. 2014;185:32-40.
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunological reviews. 2009;227(1):54-65.
- Akira S, Takeda K. Toll-like receptor signalling. Nature reviews. 2004;4(7):499-511.
- Miorin L, Albornoz A, Baba MM, D’Agaro P, Marcello A. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus research. 2012;163(2):660-666.
- Daffis S, Suthar MS, Gale M, Jr., Diamond MS. Measure and countermeasure: type I IFN (IFN-alpha/beta) antiviral response against West Nile virus. J Innate Immun. 2009;1(5):435-445.
- Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. Journal of virology. 2010;84(17):8470-8483.
- Zegenhagen L, Kurhade C, Koniszewski N, Overby AK, Kroger A. Brain heterogeneity leads to differential innate immune responses and modulates pathogenesis of viral infections. Cytokine & growth factor reviews. 2016.
- Zegenhagen L, Kurhade C, Kroger A, Overby AK. Differences in IPS-1 mediated innate immune responses between neurotrophic flavivirus infection. . Journal of Neuroinfectious Diseases. 2016;7(210).
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. The Journal of biological chemistry. 2007;282(21):15325-15329.
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. The EMBO journal. 1998;17(4):1087-1095.
- Baker DG, Woods TA, Butchi NB, et al. Toll-like receptor 7 suppresses virus replication in neurons but does not affect viral pathogenesis in a mouse model of Langat virus infection. The Journal of general virology. 2013;94(Pt 2):336-347.
- Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. Journal of virology. 2010;84(19):9760-9774.
- Aguirre S, Maestre AM, Pagni S, et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS pathogens. 2012;8(10):e1002934.
- Dalrymple NA, Cimica V, Mackow ER. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. MBio. 2015;6(3):e00553-00515.
- Miorin L, Romero-Brey I, Maiuri P, et al. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. Journal of virology. 2013;87(11):6469-6481.
- Overby AK, Weber F. Hiding from intracellular pattern recognition receptors, a passive strategy of flavivirus immune evasion. Virulence. 2011;2(3):238-240.
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nature reviews. 2008;8(7):559-568.
- Taylor RT, Lubick KJ, Robertson SJ, et al. TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell host & microbe. 2011;10(3):185-196.
- Upadhyay AS, Vonderstein K, Pichlmair A, et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cellular microbiology. 2014;16(6):834-848.
- Upadhyay AS, Stehling O, Panayiotou C, Rosser R, Lill R, Overby AK. Cellular requirements for iron-sulfur cluster insertion into the antiviral radical SAM protein viperin. The Journal of biological chemistry. 2017.
- Lindqvist R, Kurhade C, Gilthorpe JD, Overby AK. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J Neuroinflammation. 2018;15(1):80.
- Lindqvist R, Overby AK. The Role of Viperin in Antiflavivirus Responses. DNA Cell Biol. 2018;37(9):725-730.
- Lindqvist R, Upadhyay A, Overby AK. Tick-Borne Flaviviruses and the Type I Interferon Response. Viruses. 2018;10(7).
- Panayiotou C, Lindqvist R, Kurhade C, et al. Viperin restricts Zika virus and tick-borne encephalitis virus replication by targeting NS3 for proteasomal degradation. Journal of virology. 2018.
- Vonderstein K, Nilsson E, Hubel P, et al. Viperin targets flavivirus virulence by inducing assembly of non-infectious capsid particles. Journal of virology. 2017;92(1).
- Claude A, Zhao BP, Kuziemsky CE, et al. GBF1: A novel Golgi-associated BFA-resistant guanine nucleotide exchange factor that displays specificity for ADP-ribosylation factor 5. The Journal of cell biology. 1999;146(1):71-84.
- Niu TK, Pfeifer AC, Lippincott-Schwartz J, Jackson CL. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol Biol Cell. 2005;16(3):1213-1222.
- Carpp LN, Rogers RS, Moritz RL, Aitchison JD. Quantitative proteomic analysis of host-virus interactions reveals a role for Golgi brefeldin A resistance factor 1 (GBF1) in dengue infection. Mol Cell Proteomics. 2014;13(11):2836-2854.
- Lanke KH, van der Schaar HM, Belov GA, et al. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. Journal of virology. 2009;83(22):11940-11949.
- Liang W, Zheng M, Bao C, Zhang Y. CSFV proliferation is associated with GBF1 and Rab2. J Biosci. 2017;42(1):43-56.
- Zhang N, Zhang L. Key components of COPI and COPII machineries are required for chikungunya virus replication. Biochemical and biophysical research communications. 2017;493(3):1190-1196.
- Lindqvist R, Mundt F, Gilthorpe JD, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation. 2016;13(1):277.
- Best SM, Morris KL, Shannon JG, et al. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. Journal of virology. 2005;79(20):12828-12839.
- Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cellular microbiology. 2008;10(3):696-712.
- Lubick KJ, Robertson SJ, McNally KL, et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell host & microbe. 2015;18(1):61-74.
- Avirutnan P, Fuchs A, Hauhart RE, et al. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. The Journal of experimental medicine. 2010;207(4):793-806.
- Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. 2011;187(1):424-433.
- Jacobs SC, Stephenson JR, Wilkinson GW. Protection elicited by a replication-defective adenovirus vector expressing the tick-borne encephalitis virus non-structural glycoprotein NS1. The Journal of general virology. 1994;75 ( Pt 9):2399-2402.
- Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163-194.
- Dropulic LK, Cohen JI. Severe viral infections and primary immunodeficiencies. Clin Infect Dis. 2011;53(9):897-909.
- Azeredo EL, De Oliveira-Pinto LM, Zagne SM, Cerqueira DI, Nogueira RM, Kubelka CF. NK cells, displaying early activation, cytotoxicity and adhesion molecules, are associated with mild dengue disease. Clin Exp Immunol. 2006;143(2):345-356.
- Larena M, Regner M, Lobigs M. Cytolytic effector pathways and IFN-gamma help protect against Japanese encephalitis. Eur J Immunol. 2013;43(7):1789-1798.
- Strauss-Albee DM, Fukuyama J, Liang EC, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med. 2015;7(297):297ra115.
- Vargin VV, Semenov BF. Changes of natural killer cell activity in different mouse lines by acute and asymptomatic flavivirus infections. Acta virologica. 1986;30(4):303-308.
- Blom K, Braun M, Pakalniene J, et al. NK Cell Responses to Human Tick-Borne Encephalitis Virus Infection. J Immunol. 2016;197(7):2762-2771.
- Kawai T, Akira S. Innate immune recognition of viral infection. Nature immunology. 2006;7(2):131-137.
- Palmer DR, Sun P, Celluzzi C, et al. Differential effects of dengue virus on infected and bystander dendritic cells. Journal of virology. 2005;79(4):2432-2439.
- Qian F, Wang X, Zhang L, et al. Impaired interferon signaling in dendritic cells from older donors infected in vitro with West Nile virus. The Journal of infectious diseases. 2011;203(10):1415-1424.
- Cao S, Li Y, Ye J, et al. Japanese encephalitis Virus wild strain infection suppresses dendritic cells maturation and function, and causes the expansion of regulatory T cells. Virology journal. 2011;8:39.
- Robertson SJ, Lubick KJ, Freedman BA, Carmody AB, Best SM. Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signaling to inhibit dendritic cell function. J Immunol. 2014;192(6):2744-2755.
- Pierson TC, Fremont DH, Kuhn RJ, Diamond MS. Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell host & microbe. 2008;4(3):229-238.
- Kreil TR, Eibl MM. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model. Journal of virology. 1997;71(4):2921-2927.
- Heinz FX, Berger R, Tuma W, Kunz C. A topological and functional model of epitopes on the structural glycoprotein of tick-borne encephalitis virus defined by monoclonal antibodies. Virology. 1983;126(2):525-537.
- Niedrig M, Klockmann U, Lang W, et al. Monoclonal antibodies directed against tick-borne encephalitis virus with neutralizing activity in vivo. Acta virologica. 1994;38(3):141-149.
- Phillpotts RJ, Stephenson JR, Porterfield JS. Passive immunization of mice with monoclonal antibodies raised against tick-borne encephalitis virus. Brief report. Archives of virology. 1987;93(3-4):295-301.
- Kreil TR, Maier E, Fraiss S, Eibl MM. Neutralizing antibodies protect against lethal flavivirus challenge but allow for the development of active humoral immunity to a nonstructural virus protein. Journal of virology. 1998;72(4):3076-3081.
- Cerwenka A, Morgan TM, Dutton RW. Naive, effector, and memory CD8 T cells in protection against pulmonary influenza virus infection: homing properties rather than initial frequencies are crucial. J Immunol. 1999;163(10):5535-5543.
- Krylova NV, Smolina TP, Leonova GN. Molecular Mechanisms of Interaction Between Human Immune Cells and Far Eastern Tick-Borne Encephalitis Virus Strains. Viral Immunol. 2015;28(5):272-281.
- Blom K, Braun M, Pakalniene J, et al. Specificity and dynamics of effector and memory CD8 T cell responses in human tick-borne encephalitis virus infection. PLoS pathogens. 2015;11(1):e1004622.
- Fujii Y, Hayasaka D, Kitaura K, Takasaki T, Suzuki R, Kurane I. T-cell clones expressing different T-cell receptors accumulate in the brains of dying and surviving mice after peripheral infection with far eastern strain of tick-borne encephalitis virus. Viral Immunol. 2011;24(4):291-302.
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. Journal of neuropathology and experimental neurology. 2005;64(6):506-512.
- Michlmayr D, Bardina SV, Rodriguez CA, Pletnev AG, Lim JK. Dual Function of Ccr5 during Langat Virus Encephalitis: Reduction in Neutrophil-Mediated Central Nervous System Inflammation and Increase in T Cell-Mediated Viral Clearance. J Immunol. 2016;196(11):4622-4631.
- Zompi S, Harris E. Animal models of dengue virus infection. Viruses. 2012;4(1):62-82.
- Dobler G, Bestehorn M, Antwerpen M, Overby-Wernstedt A. Complete Genome Sequence of a Low-Virulence Tick-Borne Encephalitis Virus Strain. Genome Announc. 2016;4(5).
- Asghar N, Lee YP, Nilsson E, et al. The role of the poly(A) tract in the replication and virulence of tick-borne encephalitis virus. Sci Rep. 2016;6:39265.
- Kurhade C, Schreier S, Lee YP, et al. Correlation of Severity of Human Tick-Borne Encephalitis Virus Disease and Pathogenicity in Mice. Emerg Infect Dis. 2018;24(9):1709-1712.
- Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1989;1(3):285-296.
- Boyer JC, Haenni AL. Infectious transcripts and cDNA clones of RNA viruses. Virology. 1994;198(2):415-426.
- Aubry F, Nougairede A, Gould EA, de Lamballerie X. Flavivirus reverse genetic systems, construction techniques and applications: a historical perspective. Antiviral Res. 2015;114:67-85.
- Mandl CW, Ecker M, Holzmann H, Kunz C, Heinz FX. Infectious cDNA clones of tick-borne encephalitis virus European subtype prototypic strain Neudoerfl and high virulence strain Hypr. The Journal of general virology. 1997;78 ( Pt 5):1049-1057.
- Gritsun TS, Gould EA. Infectious transcripts of tick-borne encephalitis virus, generated in days by RT-PCR. Virology. 1995;214(2):611-618.
- Gritsun TS, Gould EA. Development and analysis of a tick-borne encephalitis virus infectious clone using a novel and rapid strategy. Journal of virological methods. 1998;76(1-2):109-120.
- Aubry F, Nougairede A, de Fabritus L, Querat G, Gould EA, de Lamballerie X. Single-stranded positive-sense RNA viruses generated in days using infectious subgenomic amplicons. The Journal of general virology. 2014;95(Pt 11):2462-2467.
- Mandl CW, Allison SL, Holzmann H, Meixner T, Heinz FX. Attenuation of tick-borne encephalitis virus by structure-based site-specific mutagenesis of a putative flavivirus receptor-binding site. Journal of virology. 2000;74(20):9601-9609.
- Yoshii K, Konno A, Goto A, et al. Single point mutation in tick-borne encephalitis virus prM protein induces a reduction of virus particle secretion. The Journal of general virology. 2004;85(Pt 10):3049-3058.
- Rumyantsev AA, Murphy BR, Pletnev AG. A tick-borne Langat virus mutant that is temperature-sensitive and host-range restricted in neuroblastoma cells and lacks neuroinvasiveness for immunodeficient mice. Journal of virology. 2006;80(3):1427-1439.
- Kofler RM, Hoenninger VM, Thurner C, Mandl CW. Functional analysis of the tick-borne encephalitis virus cyclization elements indicates major differences between mosquito-borne and tick-borne flaviviruses. Journal of virology. 2006;80(8):4099-4113.
- Markoff L. 5′- and 3′-noncoding regions in flavivirus RNA. Advances in virus research. 2003;59:177-228.
- Silva PA, Pereira CF, Dalebout TJ, Spaan WJ, Bredenbeek PJ. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. Journal of virology. 2010;84(21):11395-11406.
- Funk A, Truong K, Nagasaki T, et al. RNA structures required for production of subgenomic flavivirus RNA. Journal of virology. 2010;84(21):11407-11417.
- Lin KC, Chang HL, Chang RY. Accumulation of a 3′-terminal genome fragment in Japanese encephalitis virus-infected mammalian and mosquito cells. Journal of virology. 2004;78(10):5133-5138.
- Pijlman GP, Funk A, Kondratieva N, et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell host & microbe. 2008;4(6):579-591.
- Roby JA, Pijlman GP, Wilusz J, Khromykh AA. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses. 2014;6(2):404-427.
- Schnettler E, Tykalova H, Watson M, et al. Induction and suppression of tick cell antiviral RNAi responses by tick-borne flaviviruses. Nucleic Acids Res. 2014;42(14):9436-9446.
- Gritsun TS, Venugopal K, Zanotto PM, et al. Complete sequence of two tick-borne flaviviruses isolated from Siberia and the UK: analysis and significance of the 5′ and 3′-UTRs. Virus research. 1997;49(1):27-39.
- Hoenninger VM, Rouha H, Orlinger KK, et al. Analysis of the effects of alterations in the tick-borne encephalitis virus 3′-noncoding region on translation and RNA replication using reporter replicons. Virology. 2008;377(2):419-430.
- Mandl CW, Holzmann H, Meixner T, et al. Spontaneous and engineered deletions in the 3′ noncoding region of tick-borne encephalitis virus: construction of highly attenuated mutants of a flavivirus. Journal of virology. 1998;72(3):2132-2140.
- Sakai M, Muto M, Hirano M, Kariwa H, Yoshii K. Virulence of tick-borne encephalitis virus is associated with intact conformational viral RNA structures in the variable region of the 3′-UTR. Virus research. 2015;203:36-40.
- Sakai M, Yoshii K, Sunden Y, Yokozawa K, Hirano M, Kariwa H. Variable region of the 3′ UTR is a critical virulence factor in the Far-Eastern subtype of tick-borne encephalitis virus in a mouse model. The Journal of general virology. 2014;95(Pt 4):823-835.
- Asghar N, Lindblom P, Melik W, et al. Tick-borne encephalitis virus sequenced directly from questing and blood-feeding ticks reveals quasispecies variance. PLoS One. 2014;9(7):e103264.