
Chapter 5:
TBE in adults
Johannes P. Borde and Joanna Zajkowska
Key Points
- TBE is the most important tick-borne arbovirus disease of humans. Epidemiological data indicate a trend towards an increasing severity with higher age.
- A number of possible genetic and non-genetic risk factors have been identified, which might have an impact on the manifestation and severity of human disease.
- Different TBEV strains seem to cause differing clinical courses of disease. While the TBE-EU mainly causes a biphasic course, the clinical course of TBEV-FE and TBEV-SIB are mainly monophasic.
- The diagnosis of TBE is based on serological tests.
- So far there is no effective treatment of TBEV infections.
Introduction
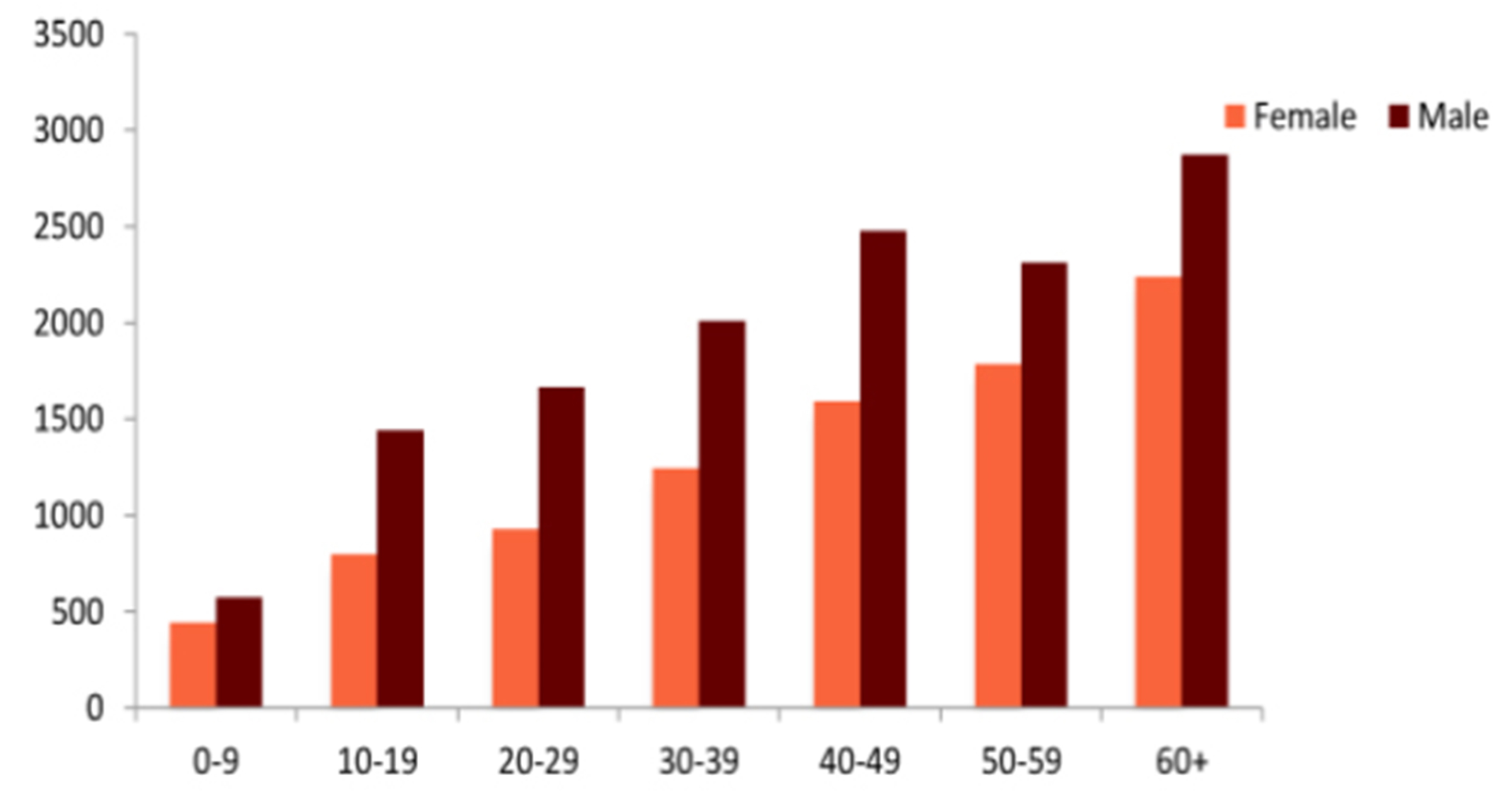
The tick-borne encephalitis virus (TBEV) causes serious infections of the brain, the myelon, and / or the meninges. Initial symptoms include headache and fever. Severe forms of tick-borne encephalitis (TBE) progress to a loss of consciousness, coma, and even death (Figure 1).1,2 Overall, TBE is associated with a high burden of disease and often disabling long-term sequelae. From an epidemiological perspective all age groups are at risk for TBE; however, the individual risk depends on age, sex, occupation, leisure activity profile, and the local environmental presence of TBEV. Within the European Union (EU), TBE became a notifiable disease in 2012, and in 2014, 2,057 cases were reported to the European Centers for Disease Control and Prevention (ECDC), 1,986 with a confirmed TBEV infection based on ECDC diagnostic criteria (0.42 cases per 100,000 population).3 The highest rates were documented in the Baltic states and TBE was predominantly reported in males years of age. In all age groups, the majority of cases occurred in males, based on aggregated data from 2000–2010 (n=22,378); however, in Lithuania, Latvia, and Estonia in the years group, female cases predominated. The ECDC report showed that most TBE cases were reported to public health authorities between June and October.3
Figure 1: TBE by age and gender
Click the image above to enlarge
Source ECDC (TBE technical report).3
Predisposition factors and risk factors
Risk factors – age, comorbidities
The personal risk for TBE in non-immune persons depends on the probability of exposure to TBEV. Exposure is usually the result of close contact with the environment in which TBEV-infected ticks are found. Forest workers, professional hunters, and landscape gardeners are examples for occupational groups with a relatively high work-related risk of acquiring a TBE infection.
However, the most endangered groups for severe clinical manifestation are elderly people, with studies showing that TBE tends to be more severe in the elderly.4 This may be due to their social habits and leisure activities, e.g., collecting mushrooms, hiking, or other activities in the forests, that lead to a higher probability of exposure to ticks and therefore for TBEV infection.
Epidemiological data from different European countries demonstrate that the people than in younger age groups. In some countries (e.g., Germany, Austria, Sweden, Lithuania) more than 50% of TBE patients are years of age. Long-term data (1993– 2008) from a single center in Białystok/Poland showed that among 710 hospitalized patients with TBE, 235 patients (33%) were years old. The clinical manifestation of TBE depends on the virulence of the pathogen and individual factors of the host. The most important host risk factors are age, comorbidities, and notably immune-suppression. In detail, the latter encompass autoimmune diseases such as rheumatoid arthritis, psoriasis, vasculitis, encephalomyelitis disseminata, solid organ recipients, or oncological diseases requiring treatment with biologicals, steroids, chemotherapeutic agents, or other immunomodulatory substances. The case fatality rate for TBE is exceedingly high in these patient groups. A recently published cluster of TBEV in the organ transplant recipients underscores the role of host immune suppression and fetal outcome.5
Another factor that may result in a more severe clinical picture of TBE is the relatively rare occurrence of coinfection with other tick-borne pathogens like Borrelia burgdorferi, Anaplasma , Rickettsia spp. or Listeria monocytogenes.6 According to Pikelj et al., other predictors for severe courses of TBE infection are early alteration of consciousness (Glasgow Coma Scale [GCS] score <7), development of limb paralysis together with respiratory insufficiency within 24–48 hours from the beginning of the second phase of the disease, and pleocytosis >300 cells.7 According to Mickiene et al., a monophasic course of the disease is also a predictive factor.8 Late onset of specific anti-TBEV IgM antibodies in cerebrospinal fluid (CSF) is linked with the severity of acute encephalitis symptoms. Thus, it can be concluded that a fatal outcome of TBE may be a consequence of coexisting risk factors, such as old age and chronic underlying diseases. Special efforts should be made to vaccinate elderly people (>50 years of age), who constitute the main group at risk for development of severe courses of TBE and long-term sequelae.6
Host genetic risk factors
As mentioned above, clinical and epidemiological data indicate that human susceptibility to clinical TBEV infection greatly varies according to age, gender, and ethnicity. The potential role of a genetic background for TBE was investigated in mouse models, where different mouse strains differ greatly in susceptibility, virus replication rate, and characteristics of the immune/inflammatory response.9 Most of the genetic factors analyzed are part of the innate immune response of the mammalian host to TBEV. A selection of genetic predispositions is discussed in the following section in the context of TBE.10
C-C chemokine receptor type 5 (CCR5)
The CCR5 plays a key role in leukocyte migration and attraction. In human immunodeficiency virus (HIV) infections, the CCR5Δ32 mutation is important for the invasion of CD4 cells by HIV particles with a CCR5 tropism. Based on these findings, the CCR5 entry inhibitor, mavaviroc, was successfully introduced into clinical use in 2007. In mouse models for flaviviral infections, it had been demonstrated that homozygous CCR5-deficient (-/-) mice died in almost 100% of all infections with West Nile virus (WNV)11; whereas CCR5 (-/+) heterozygous mice, and homozygous mice with a wildtype CCR5 receptor, had a significantly lower mortality rate after WNV infection. These data from animal studies were later confirmed in a cohort study from North America during a WNV outbreak. In conclusion, the CCR5Δ32 mutation is a strong predictor for a severe clinical course of WNV infections in the human host. Following the epidemiological results from WNV research, a potential effect of the CCR5Δ32 mutation on TBE was investigated. A clinical study from Lithuania analyzed the incidence of the CCR5Δ32 mutation in different patient populations.12 In adult patients with TBE, 2.3% (n=129) of patients were homozygous for CCR5Δ32. Control patients with aseptic meningitis (n=76), and a second healthy control group (n=134), included no patients with a homozygous CCR5Δ32 mutation. A second study with adult and pediatric TBE patients13 enrolled 246 patients in total (n=117 pediatric, n=129 adult). There were 2 control groups: 1 with 79 patients with aseptic meningitis and a second with 135 healthy individuals. Patients with TBE were stratified into subgroups on the basis of clinical parameters and severity of disease. It was shown that, in the TBE patients, a homozygous CCR5Δ32 mutation was detected with significantly greater frequency than in the control populations, but there was no correlation between disease severity and CCR5Δ32 mutational status. Mutations in another C-C chemokine receptor, CCR2, are linked to a slower progression of HIV infections; however, any association between CCR2 and TBE has not yet been studied.
Toll-like receptor 3
Toll-like receptor 3 (TLR 3) is an intracellular pattern recognition receptor (PRR), active within the cell and located to a lesser degree at the cell surface, which recognizes double-stranded ribonucleic acid (dsRNA), thus making it an important factor in the innate response to most viruses.14,15 Activation of TLR3 results in activation of the NFκB pro-inflammatory transcription factor pathway, and synthesis of type 1 and type 3 interferon (IFN).14,16 TLR3-dependent signaling has been described in different cell types, including epithelial and endothelial cells, fibroblasts, different populations of mononuclear leukocytes, as well as neurons and glial cells.14,16-19 TLR3 is present in different types of glial cells within the central nervous system (CNS) and its expression is upregulated during CNS inflammation. However, some viruses seem to suppress or bypass TLR3 signaling, at least in some types of cells. For example, herpes simplex virus type 1 (HSV1) induces only a limited response in human neurons expressing TLR3, with no type 1 IFN synthesis.19
Data from WNV infections suggest that there is a link between genetic risk factors and severe clinical courses; however, findings are, in part, inconsistent. WNV entry into the CNS depends on the expression of TLR3 as an important cofactor.21 Knockout of TLR3 results in higher peripheral viral load, reduced CNS viral load, and inflammation of the brain.21 The detrimental effects of TLR3 stimulation depend on the activation of tumor necrosis factor-α (TNF-α) receptor 1, suggesting that TNF-α is a downstream mediator of TLR3-induced blood–brain barrier permeability.21 However, knockout of TLR3 reduces the inflammatory response and thus neuronal damage in the CNS, irrespective of the local viral load.21
For TBEV, the exact mechanism of entry to the CNS is not known.22-24 It is speculated that, very similar to WNV, TBEV enters the CNS after an intense peripheral inflammatory response has disrupted the blood–brain barrier, or via olfactory neurons.25 Investigating the clinical relevance of these findings from animal studies, a clinical study analyzed the frequency of 2 TLR3 mutations in 128 TBE patients from Lithuania,26 presumably infected with the European subtype of TBEV (TBEV-EU). There were 2 control groups: 1 consisted of 77 patients with aseptic meningitis and the second comprised 135 healthy individuals from the same region. Results indicated that fully-functioning TLR3 is a risk factor for TBE. These findings were confirmed by a large cohort study from Lithuania13 and a trial from Russia.27 In the Lithuanian cohort study, which enrolled adult and pediatric TBE cases, the TLR3 polymorphism, rs3775291, was found to be less prevalent in the combined adult/pediatric TBE cohort than in the healthy control group.13 In the Russian analyses, which investigated 137 non-vaccinated TBE patients for the presence of the rs3775291 poly-morphism compared with a healthy control group (n=239), it was concluded that a functioning, wild type TLR3 seems to be a risk factor for TBE.
2’-5’ Oligoadenylate synthetase (IFN-induced oligoadenylate synthetase 2 (OAS2) and IFN-induced oligoadenylate synthetase 3 (OAS3))
2’-5’ Oligoadenylate synthetase is an IFN-induced antiviral protein that catalyzes oligomerization of ATP into 2’,5’- linked oligoadenylate (2-5A), which in turn activates latent RNase L. Activated RNase L is able to degrade viral RNA, particularly RNA from neurotropic viruses.19 The human OAS gene family comprises 4 genes, of which 3: OAS1, OAS2, and OAS3 encode functional OAS, which by alternative splicing may be expressed in 8 different isoforms: 5 of OAS1 (p42, p44, p46, p48, and p52), 2 of OAS2 (p69 and p71), and a single OAS3 isoform (p100). OASL encodes 2 isoforms of the OASL protein (p30, p59) which lacks OAS enzymatic activity. OAS1 and OAS2 synthesize mainly 2-5A oligomers and OAS3 mainly (but probably not exclusively) dimers, which do not activate RNase L, but may initiate alternative antiviral pathways. Particular isoforms of OAS1 and OAS2 differ in their activity and intracellular distribution.28 Constitutive OAS activity is individually variable and strongly correlated between close relatives, suggesting that genetic factors determine activity. In theory, single nucleotide polymorphisms (SNPs) in all the genes of the OAS complex could contribute to the phenotypic variability, as well as different isoforms of OAS proteins, contributing to the overall activity in vivo. Several findings from other viral infections point to an important role of OAS genes in the anti-flaviviral immune response. For example, in patients with dengue shock syndrome, transcription levels of OAS3 in peripheral blood mononuclear cells are significantly lower than in patients with the less severe form of the disease.30 The very initial stage of infection with TBEV depends on virus replication in the Langerhans cells, macrophages, and neutrophils in the skin at the site of tick feeding.31 It is worthy of speculation that the innate antiviral response, including OAS activity, could influence the outcome of the infection at an early stage, protecting some individuals from clinically overt disease even before an adaptive immune response and seroconversion, very similar to the suggestion by Lim et al. in the context of WNV infection.28,31 There are few clinical data regarding TBE and mutations in the OAS genes. In a study by Kindberg et al., rs1077471 distribution was not significantly different between healthy controls and TBE patients from Lithuania, although there was a non-significant tendency towards a more frequent homozygosity for the mutant allele in meningoencephalitis patients (7% in TBE group vs. 11% in non-TBE group) than in healthy individuals (3%).26 Barkhash et al. studied 23 SNPs within the OAS gene cluster in a group of patients from Novosibirsk, presumably infected with the Siberian TBEV subtype (TBEV-Sib), and identified 3 SNPs in the OAS2 gene (rs1293762, rs15895, and rs1732778) and 2 SNPs in OAS3 gene (rs2285932, rs2072136). There were significant differences in allele and haplotype frequency of these SNPs between patients with mild TBEV infection (uncomplicated meningitis and febrile disease) and with encephalo-meningits or myelitis. However, in contrast with other studies,26,28 no SNPs in the remaining genes of the OAS cluster (OAS1 and OASL) were associated with the severity of infection. The authors of the Russian study analyzed, in a second step,27 the frequency of such OAS ‘TBE risk SNPs’ in different ethnic populations within the territory of the Russian Federation. The authors found that the frequency of the aforementioned SNPs correlated with the probability of exposure to TBEV. Very low SNP frequencies were detected in Altaians, Khakasses, Tuvinians, and Shorians, groups that have a high exposure risk for TBEV in their native habitats. In conclusion, these ‘TBE risk SNPs’ may even serve as selection factors in these ethnic groups.
IL-28 and IL-10 polymorphisms
The IL-28B polymorphism has been used in the era before the ‘directly acting antivirals’ (DAA) revolution to predict a sustained virological response (SVR) in patients chronically infected with the hepatitis C virus (HCV). The IL- 28B polymorphism (rs12979860) is associated with an improved SVR in response to an antiviral HCV regimen based on pegylated IFN, proteinase-inhibitors, and optional ribavirin.32 Given the close genetic relationship of flaviviral pathogens like HCV and TBEV, the role of the IL-28B and IL-10 polymorphism was investigated in TBEV infections.33 In a study from the Novosibirsk region of Russia, 132 non-vaccinated patients with TBE were compared with a regional control group comprising 221 healthy individuals. The results indicated that the IL-28B polymorphism (rs8103142, rs12980275) and the IL-10 polymorphism (rs1800872) are genetic risk factors for TBE and in particular for severe TBE disease.33
CD209 – (ICAM)-3-grabbing non-integrin (DC-SIGN)
Dendritic cell (DC)-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) is a C-type lectin, expressed by DCs and a subpopulation of macrophages, involved in detection of pathogen-associated molecular patterns (PAMPs), cell migration, and interaction with T lymphocytes, potentially contributing to an early response to TBEV at the site of tick feeding and initiation of a specific immune response. It is coded by CD209 gene on the chromosome.19 Findings in the context of dengue virus and HCV infections pointed to an increased risk of dengue hemorrhagic fever and advanced hepatic injury in hepatitis C when there is an underlying SNP (rs4804803) located in the promotor region of the CD209 gene.34 Dendritic cells in the skin and gut probably play an important role in the early stages of TBEV infection. These antigen-presenting cells act as a source of pro-inflammatory and antiviral mediators and as initiators of a specific immune response. However, this cell type is susceptible to TBEV as well and facilitates the initial spread of TBEV from the primary site of infection to remote regions of the mammalian host.31 A clinical study from Russia enrolling 136 non-vaccinated TBE patients showed a correlation between the presence of 2 SNPs (rs4804803, rs2287886) in the region of the CD209 gene and severity of TBE disease course.34 The studied patient population was stratified into different clinical syndromes–isolated febrile illness (n=35), meningitis (n=61), and severe CNS manifestation such as meningoencephalitis (n=40). The control group comprised 263 healthy individuals from the same Siberian region. The Russian TBE cases were presumably infected with the TBEV- Sib. However, it should be noted that, to date, there has been no study investigating CD209 polymorphisms in European TBE patients infected with the western subtype of TBEV.
Clinical course
Pathogenesis – preclinic phase
After a tick bite, the released TBEV in the skin replicates subcutaneously and is, in this early stage, limited to the skin. Dendritic cells (DC) of the skin, the Langerhans cells, bind with antigens and subsequently induce an immune response by producing proinflammatory cytokines. After the initial replication in the skin, TBEV is brought to and then replicates in the lymph nodes and lymphatic system, leading to viremia. DCs and macrophages are crucially involved in TBEV replication, and may contribute to the spread to uninfected cells, thereby serving as an important source of local virus replication before viremia occurs.35,36
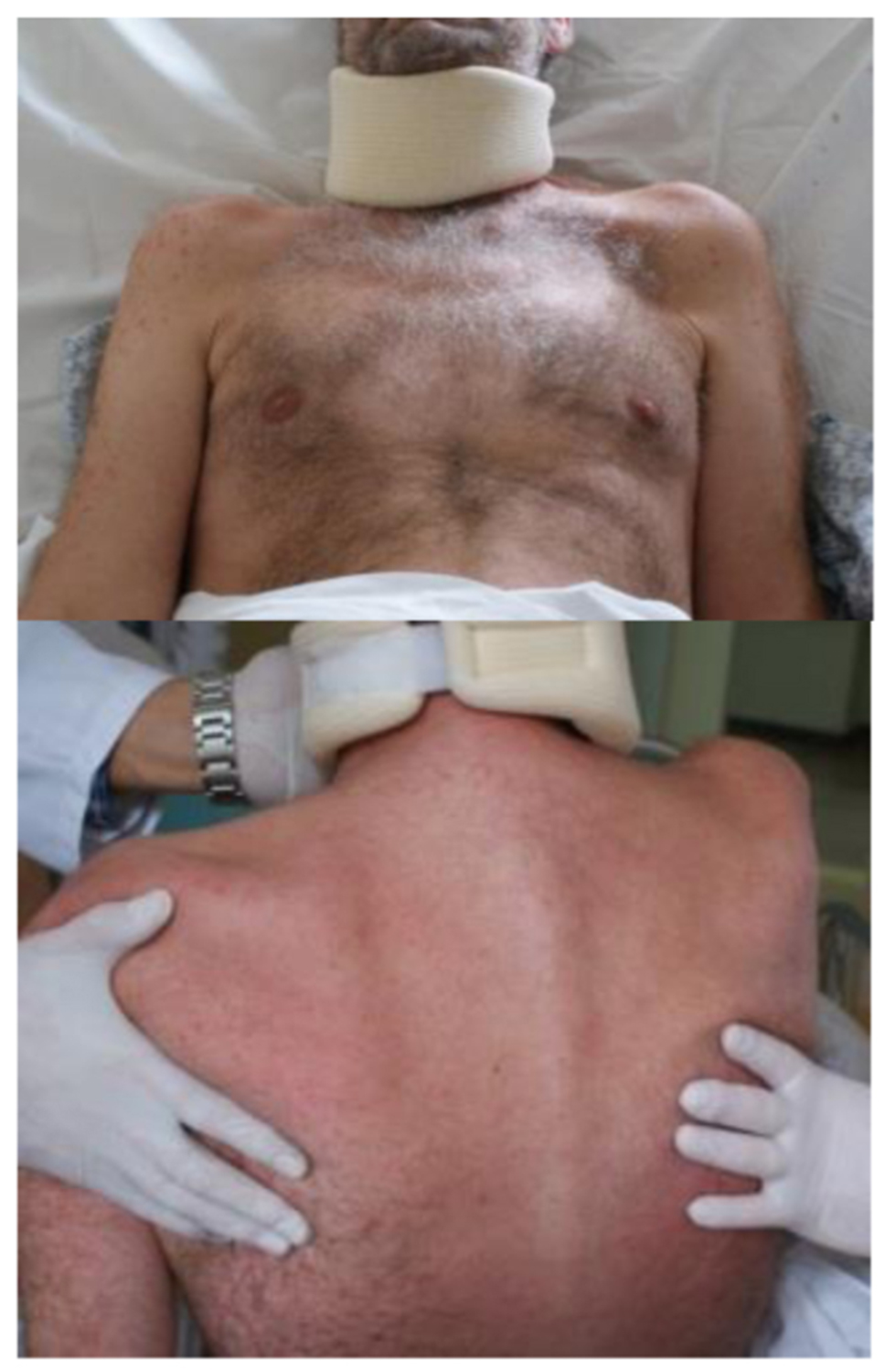
Photo 1
Click the image above to enlarge
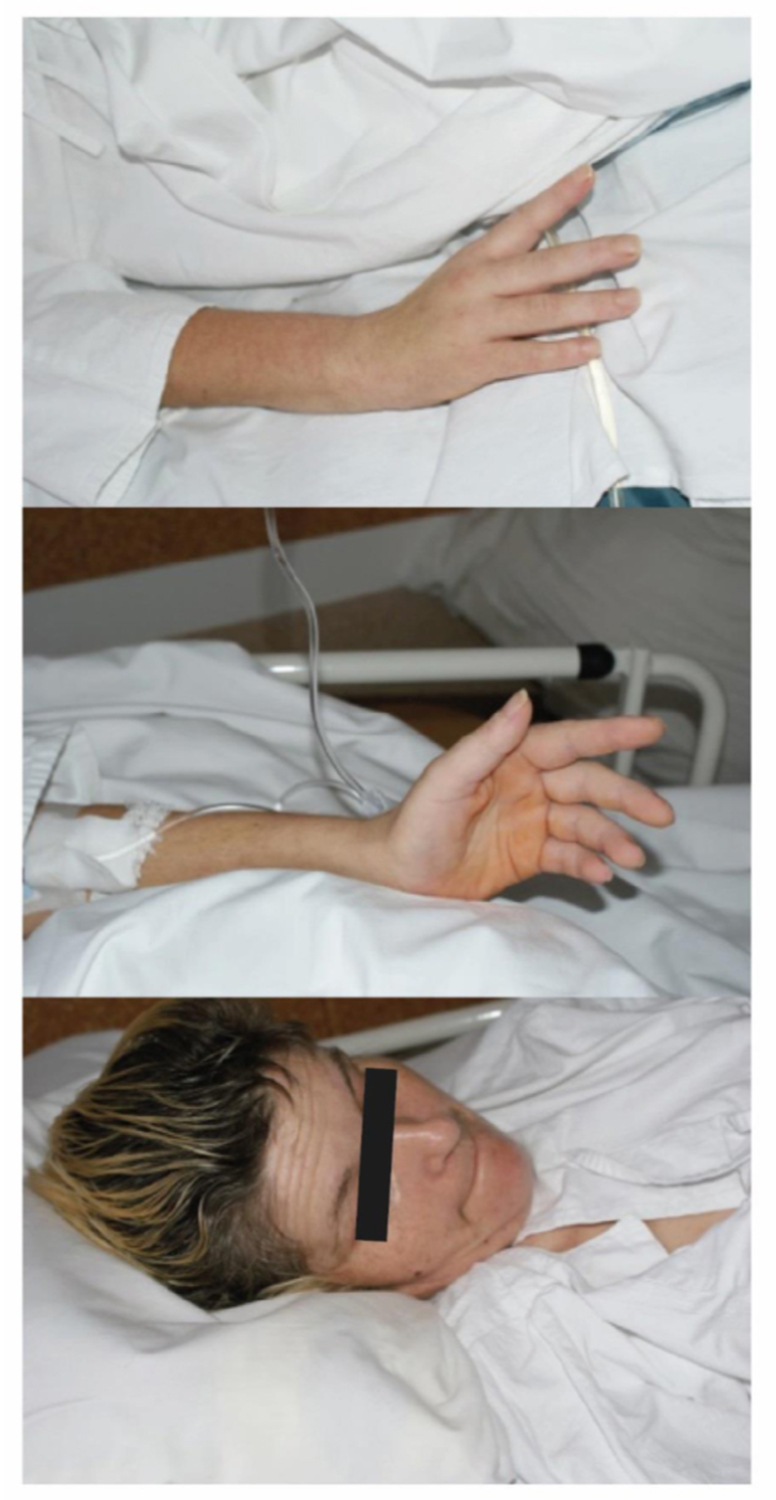
Photo 2
Click the image above to enlarge
Photo 3

Click the image above to enlarge
Time frames of the clinical presentation and definitions
The incubation period is difficult to assess in many TBE cases, because tick bites often remain unnoticed. There are few data and only rough estimates about the percentage of infected individuals who develop symptomatic disease.2 Published data suggest an average incubation period of 7–10 days (range, 4–28 days) after a bite of an infected tick.8,37
In a recent Polish study, a median incubation period as long as 22 days (range, 4-34 days) has been reported 38 However, in a case of food-borne TBEV transmission, the incubation period may be even shorter and depends upon the number of viable virus particles ingested with food. 3-4 days have been reported by Hudopisk et al.39 and by Dumpis et al.40 in a recent outbreak of alimentary TBE in Austria, the length of the incubation period ranged between 9 and 14 days.41
Symptoms of TBEV infection usually appear in a 2-phase course. In order to harmonize the use of terminology, TBEV infection is categorized into a first phase, which progresses to a second (neurological) phase of the disease. Sometimes, the disease progression is terminated after the first phase; this clinical pattern is termed ‘abortive’. Monophasic disease expresses only 1 phase of the disease with neurological symptoms. The typical course of the infection exhibits the aforementioned 2 phases and is called ‘biphasic’. The TBEV-Sib might even cause chronic disease.38,39
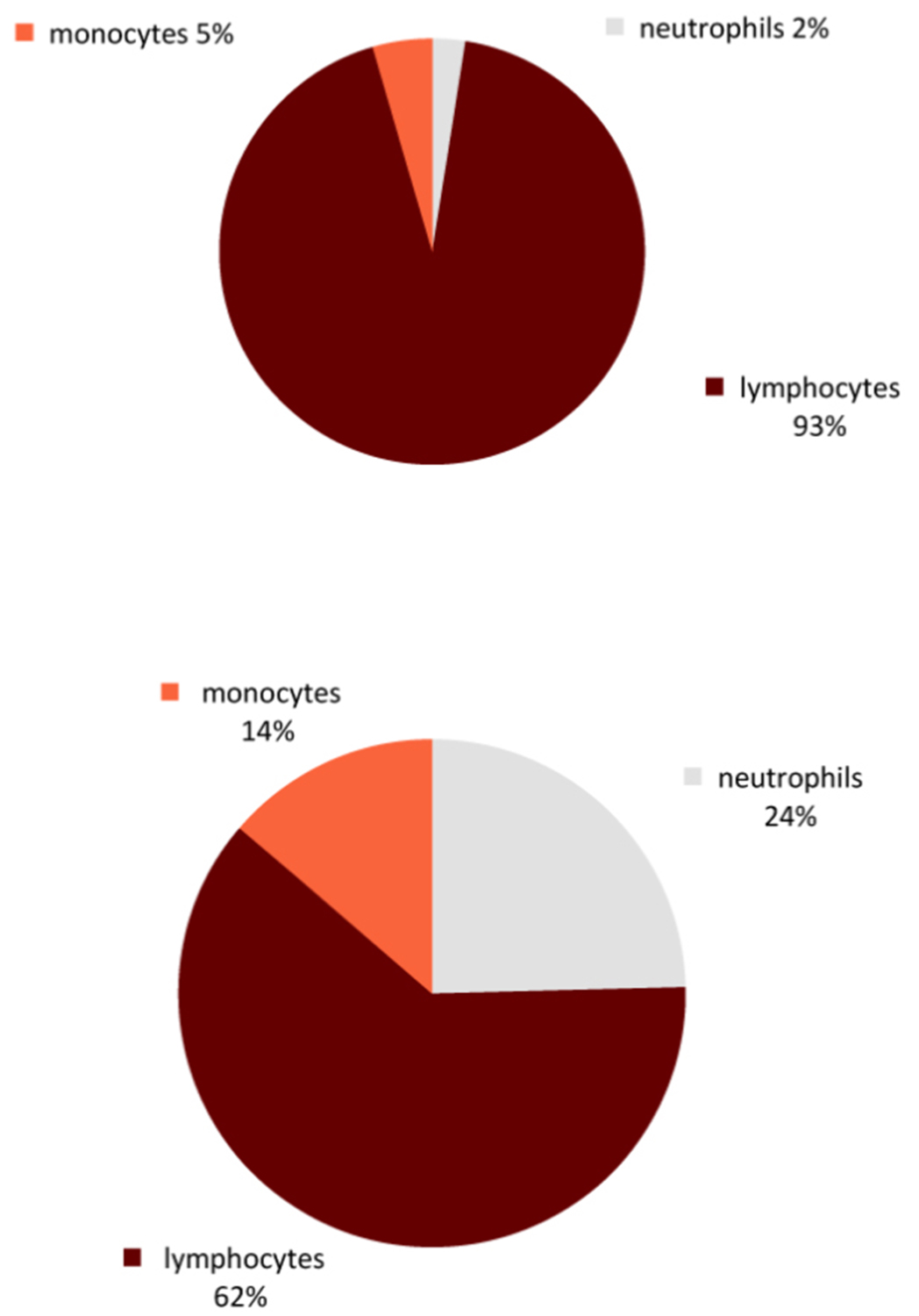
Figure 2: Pleocytosis in CSF
Click the image above to enlarge
Dynamics of TBE – first phase and second phase
During the viremic phase, extra-neural tissues, including the reticuloendothelial system (spleen, liver, and bone marrow), are infected later and release TBEV. Viremia lasts for several days and facilitates the invasion of the CNS.40 The outcome at this stage depends on the initial immune response at the entry site and the subsequent peripheral immune response. Impaired clearance of viremia and consequent entry of TBEV to the CNS are the result of a limited humoral response in the early course of TBEV infection.25 Flu-like symptoms develop during the initial viremic phase of the illness, including fever, headache, fatigue, myalgia, anorexia, nausea, and vomiting. However, fever and headache are the chief complaints of patients presenting to health services. This initial phase lasts for 2–4 days (range 1–8 days).41,42
Table 1: Overview of TBE long-sequelae in prospective and retrospective studies
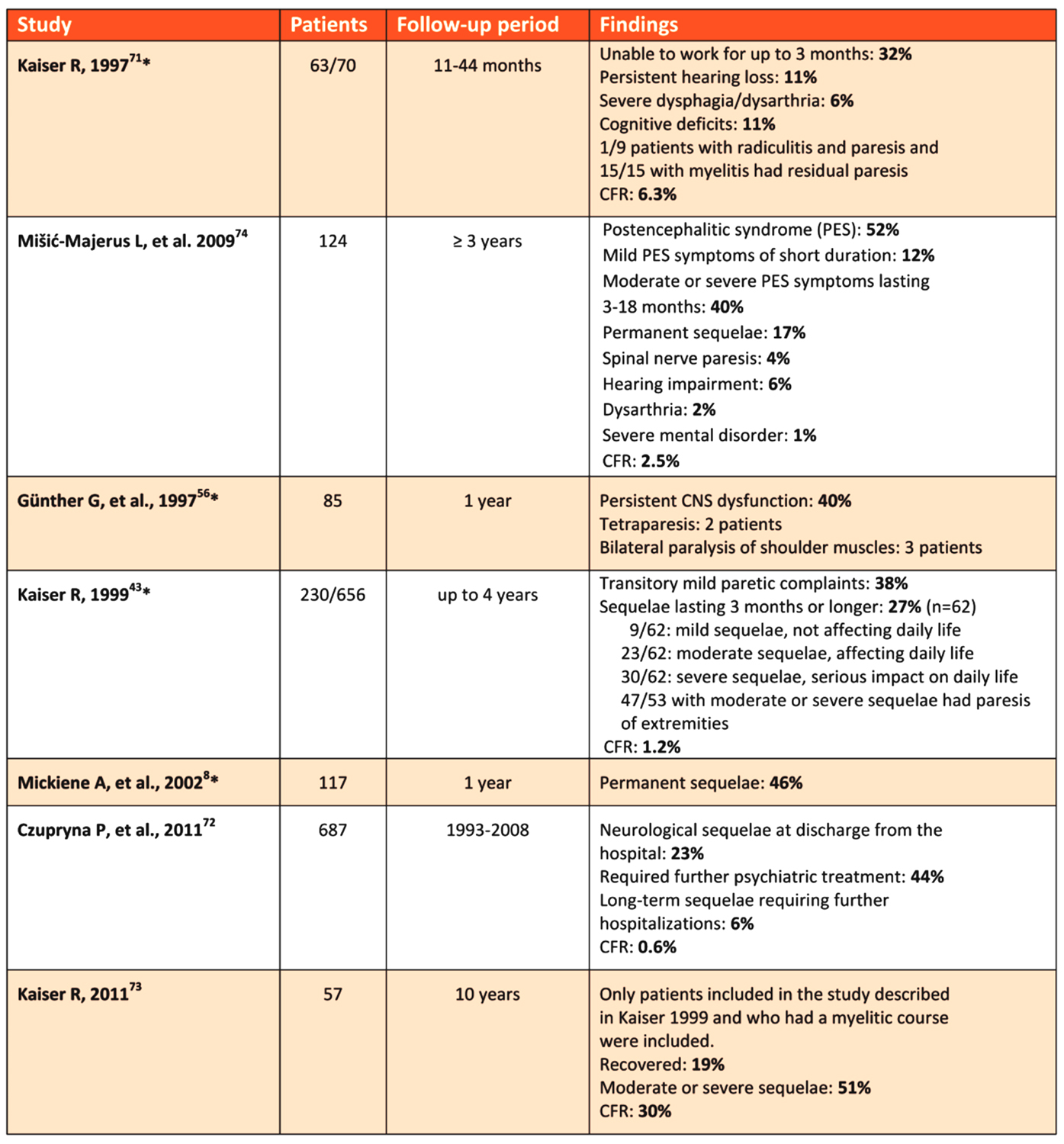
Click the image above to enlarge
Overall, approximately of all infected individuals remain free of clinical signs and symptoms, and of all symptomatic TBE patients experience only the initial phase; however, epidemiological data show wide variability. An estimated of patients with TBE who have gone through the initial phase will develop a second phase with CNS involvement. The asymptomatic period between the first and second phase of symptoms lasts for 8 (1–20) days.41,43 Abortive disease is diagnosed if there is only an initial phase. The second phase of the disease may involve the CNS with symptoms of meningitis, meningoencephalitis, meningoencephalomyelitis, encephalo-radiculitis, or even mixed forms. A biphasic course is observed in 74–85% of TBE patients infected with TBEV-EU.42 Up to 46% of patients experiencing the second phase of TBE develop long-term sequelae. Interestingly, Kaiser et al. report that patients with encephalomyelitis often present with only 1 phase (i.e., absence of prodromal phase).41 Infections with the TBEV-Sib and the Far Eastern subtype of TBEV (TBEV-FE) are predominantly monophasic. Only a small remainder shows a biphasic or even chronic pattern (8–21%).44 An abortive form of TBEV infection presenting exclusively with febrile temperatures, without CNS involvement, has been reported in some European studies.45 This abortive pattern seems to be a rare clinical manifestation, estimated to account for 1.8% of all TBEV infections in Europe.46 In Russia, however, these abortive forms represent up to 50% of all clinical presentations of TBE.39 The exact ratio of abortive versus CNS forms of TBE still remains unknown and depends on a variety of pathogen-related and individual host factors.8
Seroconversion without any obvious clinical signs is common and well documented, especially in populations that are highly exposed to TBEV-infected ticks.47 In a study from Sweden,48 25% of infected individuals developed CNS disease. This proportion seems to be lower with the TBEV-Sib and TBEV-FE subtypes, where 70–95% of infections are reported to remain without any symptoms.49
Clinical presentations of the neurological (second) phase
The exact mechanism by which TBEV crosses the blood–brain barrier and invades the CNS remains unclear. There are 4 proposed mechanisms: I. Cytokine mediated (targeting the endothelium) whereby cytokines modulate endothelial cell permeability, disrupt the blood–brain barrier, and lead to the passage of the TBEV into CNS; II. Trojan horse hypothesis, in which immune cells migrate into CNS and establish an infection of the neural cells, endothelial cells, or choroid plexus epithelial cells, with budding of TBEV into the parenchymal compartment;22-23,50-51 III. Digestive tract infections from epithelial cells to DCs; and IV. Infection via olfactory epithelium and olfactory neurons.
Figure 3: Evaluating pleocytosis in TBE (early)
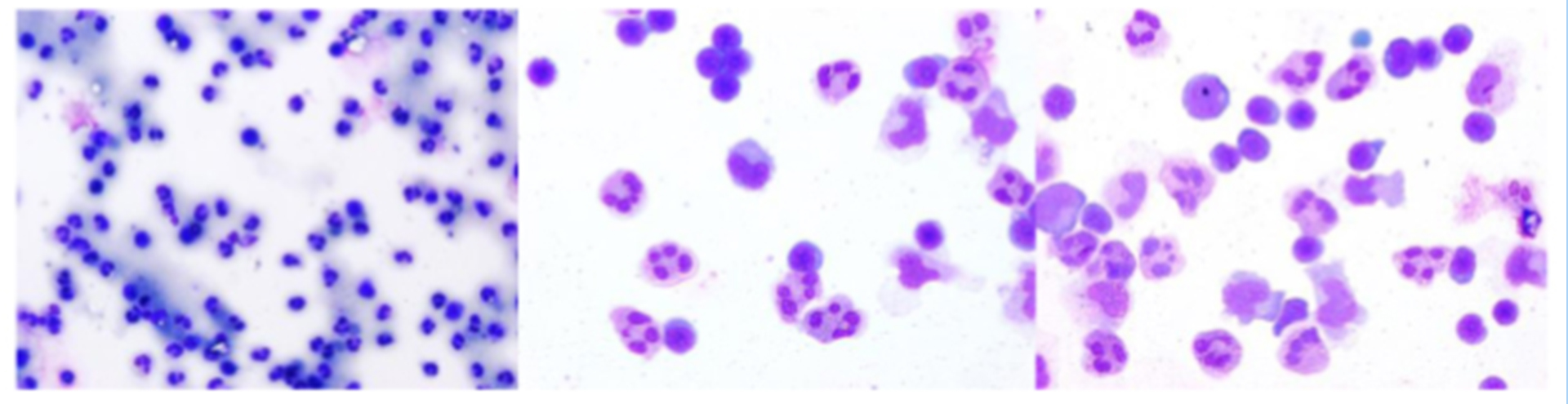
Click the image above to enlarge
(1 x 100; 1 x 400; 1 x 400.)
Figure 4: Evaluating pleocytosis in TBE (later)
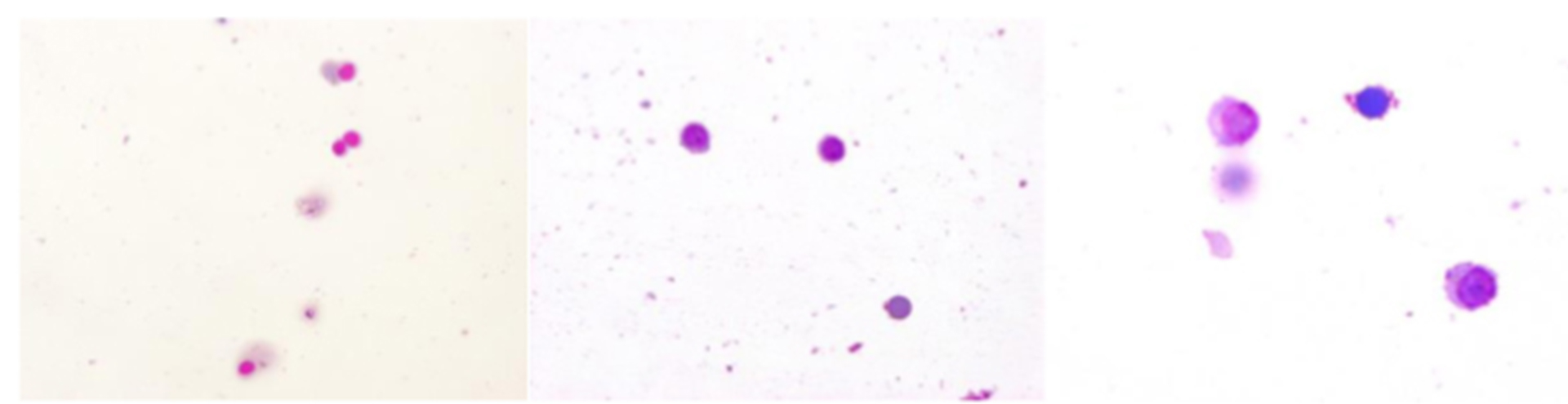
Click the image above to enlarge
lack of granulocytes x 200 x 400.
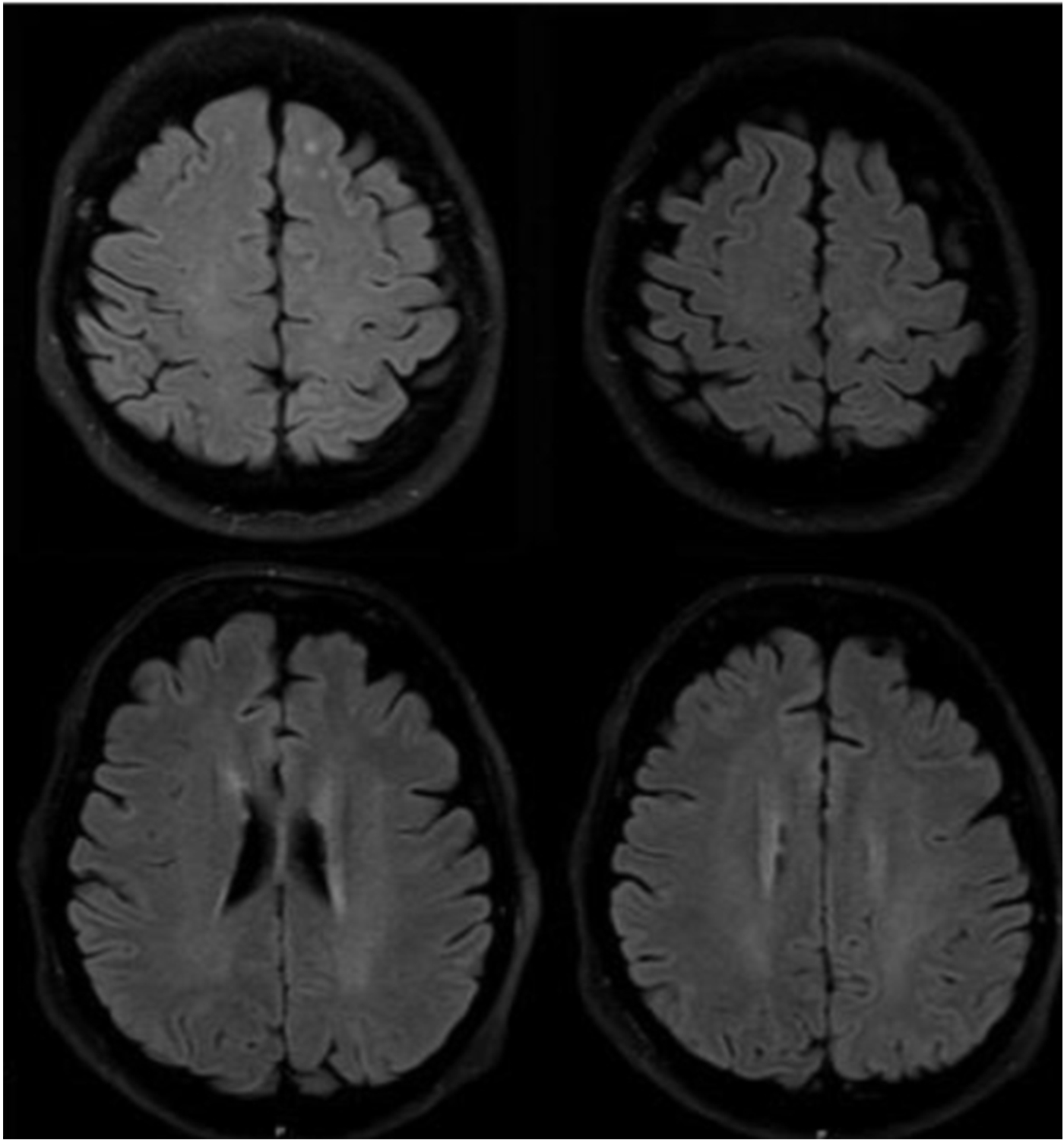
Figure 5: MRI visualization of TBE-related abnormalities
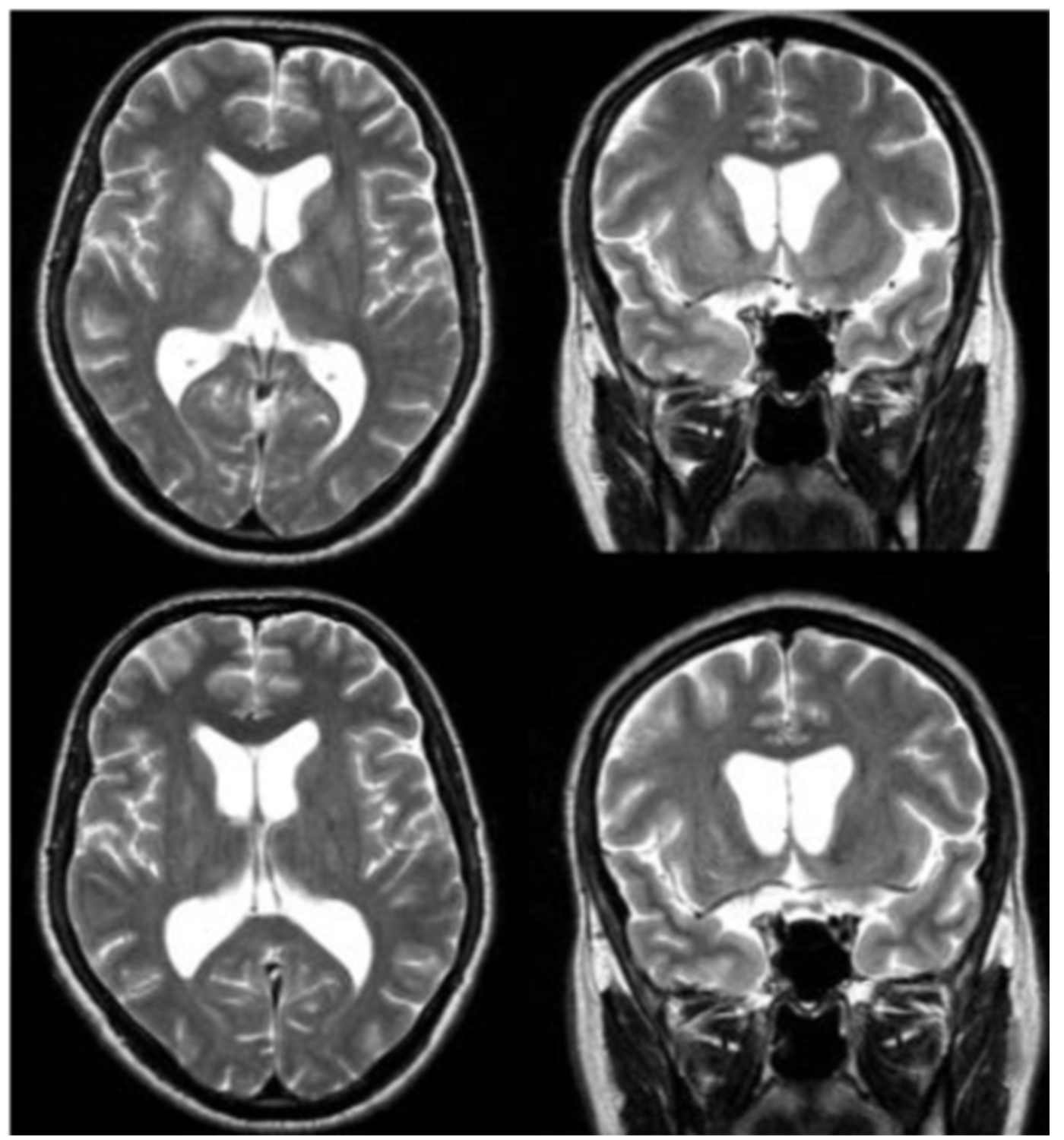
Click the image above to enlarge
Figure 6: MRI visualization of TBE-related abnormalities
Click the image above to enlarge
Figure 7: Additional visualization of TBE-related abnormalities
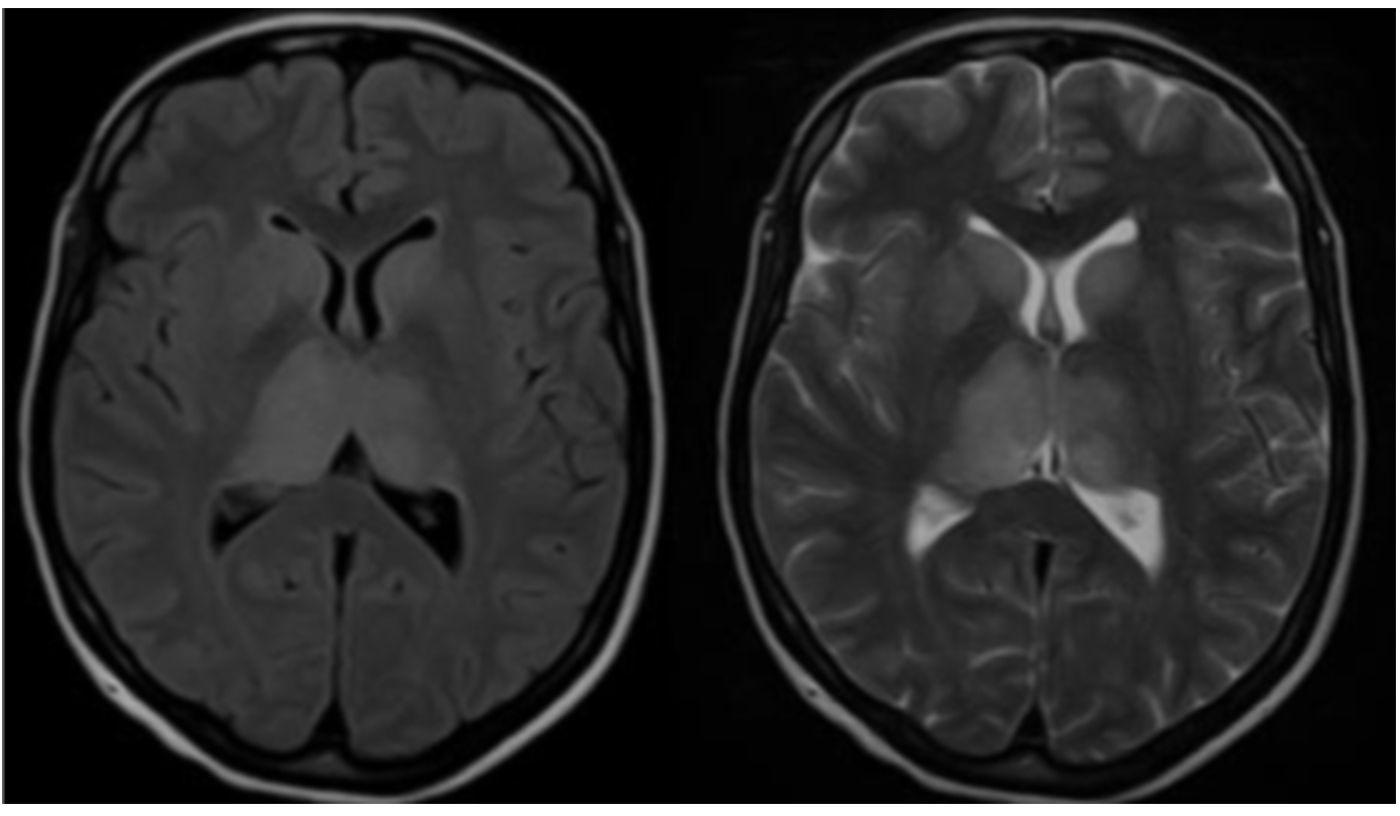
Click the image above to enlarge
The second stage of TBE begins with an increasing body temperature. This second febrile phase is characterized by temperatures 1–2°C higher than peak body temperatures in the first phase, frequently exceeding 40°C.41 The further course of acute TBE can be classified as mild, moderate, or severe depending on the affected parts of the CNS, such as meninges (meningitis), brain (encephalitis), cranial, or spinal nerves (meningoencephalomyelitis). The specific clinical symptoms in the second stage of TBE result from the affinity of the virus for distinct CNS regions, producing additional clinical symptoms like chorea,52 parkinsonism, mutism, nystagmus, and others. Encephalitic symptoms are classified as mild to severe.
Photo 4
Click the image above to enlarge
Meningitis presents with headache, nausea, vomiting, vertigo, and neck stiffness. Signs of meningeal irritation (neck stiffness, Brudziński, Kernig’s signs) have a low clinical sensitivity and could even be absent, leaving headache and febrile temperatures as the only symptoms. In a study from Poland, 10% of the TBE patients with CSF pleocytosis were without objective meningeal symptoms.53,54 In pediatric TBE patients, fever without neuro-logical symptoms is more often the chief complaint compared with adult patients.
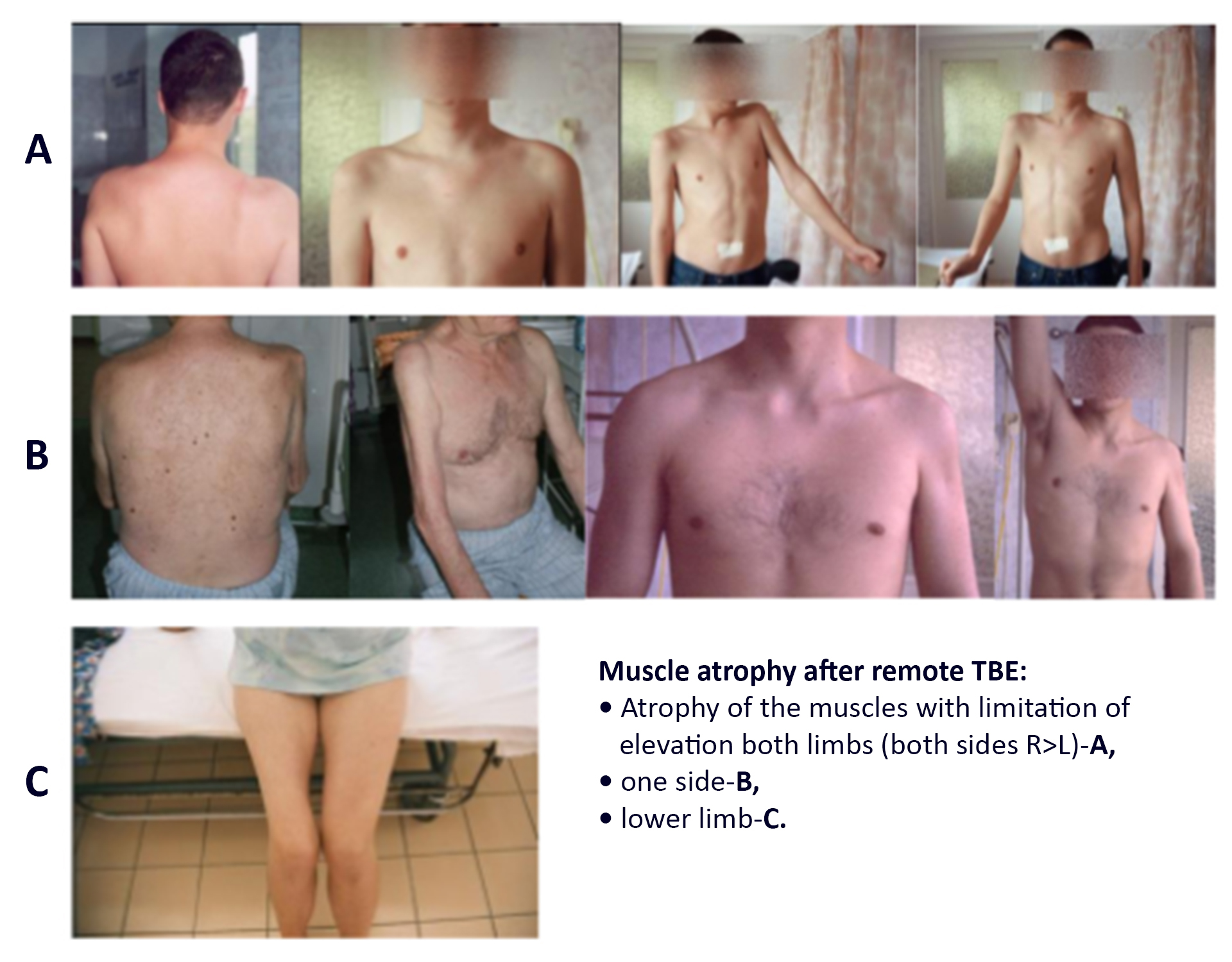
Meningoencephalitis is observed in adults in 50% of TBE cases.55,56 Symptoms are cerebellar signs and typically include ataxia. The most common neurological symptom is altered mental state, ranging from somnolence to coma with 12% of TBE patients in this phase exhibiting a GCS score below 7.41 Disorientation, excitation, seizures, and confusion are also observed, as well as hyperkinesia of limbs and facial muscles, cranial nerve involvement with paresis of facial and ocular nerves, cerebellar ataxia, and autonomic disturbances of the bladder and intestines. Spinal nerve paralysis has been documented in 11-15% of patients. Depending on the extent of the CNS affection, meningoencephalitis can be moderate or severe. Severe myalgia in the extremities sometimes precedes the development of paresis. Involvement of the cranial nerve nuclei and motor neurons of the spinal cord causes flaccid paralysis of neck and upper extremity muscles (Photo 1).
Patients with meningo-encephalo-myelitis may experience paresis of the arms, back, and legs, with the upper extremities affected more often than the lower extremities. Bilateral paresis is a rare symptom (Photo 1 and 2). Involvement of the medulla oblongata and the central parts of the brainstem (bulbar) is associated with the poor prognosis41 (Photo 3). Occasionally, TBE can be associated with autonomic dysfunction including reduced heart rate variability and tachycardia.
Flaccid paralysis arises, very similar to that seen in poliomyelitis, due to the viral preference for the anterior horn of the cervical spinal cord. In contrast to poliomyelitis, mono-, para-, or tetraparesis develops in 5–10% of patients. Paralysis of respiratory muscles may also occur, necessitating ventilatory support. Cranial nerve involvement is associated mainly with ocular, facial, and pharyngeal motor functions. Hearing defects may also occur. Brainstem involvement (particularly of the medulla oblongata) can lead to bulbar syndrome, with the risks of sudden respiratory and circulatory failure.6,7,41,42,57
Chronic TBE – chronic TBEV infection
Some peculiarities in the course of TBE are observed in Western Siberia. The onset of illness is more often gradual than acute, with a prodromal phase including fever, headache, anorexia, nausea, vomiting, and photophobia. These symptoms are followed by a stiff neck, sensorial changes, visual disturbances, and variable neurological dysfunctions, including paresis, paralysis, sensory loss, and convulsions. In fatal cases, death occurs within the first week after onset. The case-fatality rate is approximately 20%, compared with 1–2% for the European form. However, these findings may be biased by the different types of medical treatment available in Western and Eastern Europe. It is also supposed that, in contrast to the European form, the disease caused by TBEV-FE is more severe in children than in adults. Neurological sequelae occur in 30–80% of survivors, especially residual flaccid paralyses of the shoulder girdle and arms. Overall, there is little information available on the virulence of the recently described TBEV-SIB with respect to the course of disease in humans. The few systematic and unsystematic data accumulated over the past 20 years show that TBE is a chronic disease in 1–1.7% of cases. Chronic TBE affects mainly working-age people and children, often leading to their debilitation.38 The frequency of the chronic form is described as approximately 1–1.8% of patients.58
Hyperkinetic syndrome is the main manifestation of chronic TBE (86%), which presents as myoclonic hyperkinesia in paralyzed muscles, myoclonus of oral muscles, myoclonus of the oral, shoulder girdle, and abdominal wall muscles with Parkinson tremor, and spontaneous progressive form of chronic TBE (from myoclonic hyperkinesia in the arm to typical Kozevnikoff epilepsy).
Laboratory findings
Compared with other forms of viral meningitis, white blood cell counts in the CSF are low in TBE (median 60/µL, range 5–1200/µL).59 The CSF albumin is moderately increased; indeed, in some TBE cases, an increased albumin concentration may be the only pathological finding in the CSF.56 Increased CSF-to-serum-albumin ratio indicates an impaired blood–brain barrier, significant disruption of which can be observed in up to 60% of patients with TBE.59 Initially there is a predominance of poly-nuclear cells (granulocytes) in the CSF cell count; however, the immune response switches within a few days towards an increased lymphocytic cell count (Figures 2–4). In serum samples, patients with TBE display only moderate markers of inflammation. For example, CRP is marginally altered (median 3 mg/dL; range 1–60 mg/dL).41 TBE-specific IgM and IgG antibodies are already present in CSF samples at the time when patients are admitted to hospitals because of CNS symptoms.41 Meningeal signs can be absent in TBE patients with CSF pleocytosis. For microbiological confirmation of TBE please see Chapter 12.
Neuroimaging
Magnetic resonance imaging (MRI) is the standard for the evaluation of patients with any type of encephalitis, including TBE. However, only about 18% of patients with TBE have abnormalities on cranial MRI (cMRI) examination. These abnormalities are mainly located in the thalamus, the cerebellum, the brainstem, and the nucleus caudatus (Figure 5–7).57,60,61 MRI findings are bilateral or unilateral. The most characteristic MRI finding is the bilaterally increased signal intensity in T2-weighted images and in fluid-attenuated inversion recovery (FLAIR) images within the basal ganglia or thalamus. The localization of the MRI lesions corresponds with neuro-pathological findings. The cerebellum, brainstem, cerebral cortex, and spinal cord are further brain structures that may be clinically affected; however, pathological MRI enhancements are rarely seen in these locations. There is usually no restricted diffusion on diffusion-weighted imaging (DWI) in patients with TBE. Sometimes single-voxel 1H-MRI spectroscopy may show pathological patterns, indicating lactate or lipid peaks or other metabolic alterations in the otherwise unremarkably appearing basal ganglia or thalami. TBE patients with meningoencephalitis and unfavorable prognosis are more likely to present with MRI lesions; however, anecdotal reports show that even some patients with normal cMRI scans may die from TBE. It is difficult to correlate MRI results with clinical findings or even outcomes in TBE patients. However, there is one prospective cohort study from Germany that enrolled 111 TBE patients from 2004 to 2014. All patients were evaluated by high-resolution MRI. Clinical symptoms were scored using the modified RANKIN Scale at admission and at defined follow-up dates. The results indicate a strong correlation between meningo- encephalomyelitis documented on MRI and a poor outcome. Further risk factors for a worse outcome were age, male gender, and preexisting diabetes mellitus.61 Cranial computed tomography (CCT) is usually negative and not recommended for the diagnosis of encephalitic brain lesions. In some rare occasions CCT may show symmetric or asymmetric bilateral hypodensities of the thalami and/or basal ganglia.62-65 Single photon emission computed tomography (SPECT) is a highly sensitive functional imaging method for the detection of cerebral perfusion abnormalities. A SPECT study from Sweden including adult patients with TBE (n=73), and patients with other forms of meningoencephalitis (n=56), showed a decrease in regional cerebral blood flow (rCBF). However, these findings were not significantly related to the clinical course or the outcome of TBE.42,66 Thus, the significance of this reduced rCBF remains unclear. Another pilot study using 18F-FDG PET/CT in 10 patients with TBE showed that glucose hypometabolism was present in 7 out of 10 patients with TBE, reflecting neuronal dysfunction in areas prone to TBEV infiltration and responsible for the development of clinical signs and symptoms.67
EEG
In the acute stage of CNS inflammation, electro-encephalograms (EEGs) show pathological patterns. In a study by Lindquist, there were abnormal EEG findings in 77% of patients with TBE.57
An abnormal EEG correlates with the severity of the clinical condition of the TBE patient; however, there is no link between the degree of EEG pathology and clinical condition. In most cases, an initially abnormal EEG normalizes within a few weeks. The EEG may be of prognostic value when there is a persistence of pathological patterns or the appearance of new irregularities.68,69
Prognosis, long-term sequelae and Postencephalitic syndrome (PES)
The case fatality rate (CFR) from infection with TBEV-EU is reported to range between 1% and 2%.2,41 In lethal cases, death occurs within 5–10 days after the onset of neurological symptoms in the context of diffuse brain edema and bulbar involvement.
There are several retrospective and prospective studies from different countries regarding long-term morbidity of TBE patients (Table 1). Overall, there is a high proportion of patients with persistent post-TBE symptoms of different severity. However, differences in patient selection, age-specific exposure, access to medical care, and selective reporting of more severe cases may result in bias in these data and may explain, in part, the discrepancies between different TBEV subtypes.8,43,56,70-74,74
PES is a condition that includes residual (behavioral) changes following the recovery from viral or bacterial encephalitis according to the international classification for diseases. The symptoms are nonspecific and, in contrast to organic disorders, are often reversible. There may be a variety of residual neuro-logical dysfunctions, such as paralysis, deafness, or aphasia. Initially, doubts were raised whether PES after TBE exists; however, a recent study specifically assessed the incidence and characteristics of PES after TBE. Between 1995 and 2008, 124 patients aged 16– 76 years were followed for >3 years. Of these, 60 patients (48%) had no symptoms of PES; 15 patients (12%) had symptoms that were mild and of short duration; and the remaining 49 patients (40%) developed PES lasting for 3–18 months. In 15/49 patients (12%) PES was severe. The main characteristics of PES were psychiatric symptoms, balance and movement disorders, headache, general malaise, and reduced working ability.41 Kaiser et al. followed patients with meningo-encephalo-myelitis for 10 years.41,43,73 There were 57 patients with complete follow-up data, of whom 19% recovered, 51% had moderate/severe sequelae, and 30% died. The most substantial improvements were seen in the first 12 months after acute TBE. These follow-up results indicate that the chance for clinical improvement reaches a maximum in the first 12 months after acute TBE and decreases substantially after the first 3 years. The severity of acute TBE correlates with long-term prognosis. By contrast, patients with complete recovery within 5 years had the lowest initial deficit measured in a standardized scoring system. Mechanical ventilation was required in 30 patients with TBE in the acute phase of the disease–14 patients died during the follow-up period (7 within the first year). Respiratory symptoms resolved completely in 14 patients with TBE. Overall, there is a correlation of disease severity and prognosis. Patients with ataxia, impaired consciousness, double vision, urinary retention, or mild paresis of only 1 extremity had the best prognosis. However, TBE patients with tetraparesis and concurrent respiratory paralysis, dysphagia, or dysarthria were among those with the highest risk for a fatal outcome.
Post-mortem examinations of deceased TBE patients and animal studies provided some explanations and insights into the neuropathological mechanisms of the disease.75,76 Viral infection of neurons causes cell lysis. There is only a limited chance for improvement of muscle paresis, because neurons have restricted regenerative capabilities. TBEV has a high affinity for cranial nerve nuclei, the cells of the anterior horn of the spinal cord, the Purkinje cells in the cerebellum, and cellular components of the thalamus. Clinical improvements achieved in patients with paresis are linked to physical exercise increasing the muscular strength of neighboring muscle groups and, to a lesser extent, to learning effects in the context of neuronal plasticity. multisegmental, the resulting neuronal muscle atrophy has little or no chance for regeneration (Photo 4).
Treatment
Treatment is mainly supportive and symptomatic. No specific antiviral therapy is currently available and approved for TBEV infections. Some antiviral agents, specific immunoglobulins, and other potentially protective substances are under investigation for their anti-TBEV efficacy in vitro and clinically77-80; however, a detailed review of these ‘pipeline’ agents is beyond the scope of this chapter. If there are clinical signs and symptoms such as status epilepticus, severe meningo-encephalitis, encephalitis, and myelitis, the patient should be admitted to an NICU (neurological intensive care unit) for further monitoring and treatment. In a large study of 709 patients with TBE in Germany, 12% of patients required intensive care and 5% required assisted ventilation.43 Maintenance of an adequate cerebral perfusion and prevention of secondary complications are the main objectives of treatment. Correct positioning, deep analgosedation, and osmotherapy (mannitol, hypertonic saline) can be considered, but only for 1–2 days and provided exclusively as boluses. However, the use of mannitol did not affect the outcome in terms of survival. In the case of an increasing intracerebral pressure and a decreasing cerebral perfusion, therapeutic hypothermia might be considered. High fever is associated with increased metabolic consumption. Antipyretics, or other physical measures like cooling blankets, or infusion of cooled fluids, should be employed to reduce body temperature effectively. The use of steroids is still a matter of debate and cannot be recommended based on current evidence.8,72 Dehydration increases the risk for cerebral infarction. Severe TBE is often accompanied by hypovolemia due to a decreased intake and a secondary loss of fluids. Hyponatremia is a common condition in patients with TBE, including the syndrome of inappropriate antidiuretic hormone secretion (SIADH), cerebral salt-wasting syndrome, and reduced sodium supplementation. Mental and behavioral disturbances, delirium, and psychotic signs and symptoms may justify treatment with neuroleptics. For seizures, the administration of benzodiazepines is recom-mended. Pain and arousal cause intracranial pressure peaks by increasing the cerebral blood flow; therefore, sedatives and careful clinical monitoring are key factors in the prevention of intracranial hypertension. Rehabilitation should be introduced as soon as possible, as early introduction of rehabilitation therapy is essential to protect muscle atrophy.
Contact:
johannes.borde@web.de
Citation:
Borde JP, Zajkowska J. TBE in adults. Chapter 5. In: Dobler G, Erber W, Bröker M, Schmitt HJ, eds. The TBE Book. 6th ed. Singapore: Global Health Press; 2023. doi: 10.33442/26613980_5-6
References
- Kaiser R. Frühsommer-Meningoe Nervenarzt. 2016;87:667-80.
- Bogovic P, Strle F. Tick-borne encephalitis: A review of epidemiology, clinical characteristics, and management. World J Clin Cases. 2015;3:430.
- Epidemiological situation of tick-borne encephalitis in the European Union and Europe Free Trade Association countries. ECDC technical report. 2012.
- Czupryna P, Moniuszko A, Pancewicz SA, Grygorczuk S, Kondrusik M, Zajkowska J. Tick-borne encephalitis in Poland in years 1993-2008–epidemiology and clinical presentation. A retrospective study of 687 patients. Eur J Neurol. 2011;18:673-9.
- Lipowski D, Popiel M, Perlejewski K, et al. A Cluster of Fatal Tick-borne Encephalitis Virus Infection in Organ Transplant Setting. J Infect Dis. 2017;215:896-901.
- Zajkowska J, Czupryna P, Pancewicz S, et al. Fatal outcome of tick-borne encephalitis – a case series. Neurol Neurochir Pol. 2011;45:402-6.
- Pikelj F, Tomazic J, Maticic M, Socan M, Muzlovic I. Severe forms of tick-borne meningoencephalitis in Slovenia. J Infect. 1995;31:83-5.
- Mickienė A, Laiškonis A, Günther G, Vene S, Lundkvist Å, Lindquist L. Tickborne Encephalitis in an Area of High Endemicity in Lithuania: Disease Severity and Long-Term Prognosis. Clin Infect Dis. 2002;35:650-8.
- Palus M, Vojtíšková J, Salát J, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. 2013;10.
- Grygorczuk S, Osada J, Parczewski M, et al. The expression of the chemokine receptor CCR5 in tick-borne encephalitis. J Neuroinflammation. 2016;13:45.
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao J-L, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087-98.
- Kindberg E, Mickiene A, Ax C, et al. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis. 2008;197:266-9.
- Mickienė A, Pakalnienė J, Nordgren J, et al. Polymorphisms in Chemokine Receptor 5 and Toll-Like Receptor 3 Genes Are Risk Factors for Clinical Tick-Borne Encephalitis in the Lithuanian Population. Munderloh UG, editor. PLoS One. 2014;9:e106798.
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll- like receptor 3. Nature. 2001;413:732-8.
- Ranjith-Kumar CT, Miller W, Xiong J, et al. Biochemical and Functional Analyses of the Human Toll-like Receptor 3 Ectodomain. J Biol Chem. 2006;282:7668-78.
- Tissari J, Sirén J, Meri S, Julkunen I, Matikainen S. IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J Immunol. 2005;174:4289-94.
- Zhang S-Y, Jouanguy E, Sancho-Shimizu V, et al. Human Toll- like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev. 2007;220:225-36.
- Daffis S, Samuel MA, Suthar MS, Gale M, Diamond MS. Toll- like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349-58.
- Prehaud C, Megret F, Lafage M, Lafon M. Virus Infection Switches TLR-3-Positive Human Neurons To Become Strong Producers of Beta Interferon. J Virol. 2005;79:12893-904.
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013-21.
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366- 73.
- Palus M, Vancova M, Sirmarova J, Elsterova J, Perner J, Ruzek D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood- brain barrier integrity. Virology. 2017;507:110-22.
- Bílý T, Palus M, Eyer L, Elsterová J, Vancová M, Růžek D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci Rep. 2015;5:10745.
- Palus M, Bily T, Elsterova J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. J Gen Virol. 2014;95(Pt 11):2411-26.
- Dörrbecker B, Dobler G, Spiegel M, Hufert FT. Tick-borne encephalitis virus and the immune response of the mammalian host. Travel Med Infect Dis. 2010;8:213-22.
- Kindberg E, Vene S, Mickiene A, Lundkvist A, Lindquist L, Svensson L. A Functional Toll-Like Receptor 3 Gene (TLR3) May Be a Risk Factor for Tick-borne Encephalitis Virus (TBEV) Infection. J Infect Dis. 2011;203:523-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21216866
- Barkhash AV, Perelygin AA, Babenko VN, Brinton MA, Voevoda MI. Single nucleotide polymorphism in the promoter region of the CD209 gene is associated with human predisposition to severe forms of tick-borne encephalitis. Antiviral Res. 2012;93:64-8.
- Lim JK, Lisco A, McDermott DH, et al. Genetic Variation in OAS1 Is a Risk Factor for Initial Infection with West Nile Virus in Man. Rice CM, editor. PLoS Pathog. 2009;5:e1000321.
- Bonnevie-Nielsen V, Leigh Field L, Lu S, et al. Variation in Antiviral 2′,5′-Oligoadenylate Synthetase (2’5’AS) Enzyme Activity Is Controlled by a Single-Nucleotide Polymorphism at a Splice-Acceptor Site in the OAS1 Gene. Am J Hum Genet. 2005;76:623-33.
- Simmons CP, Popper S, Dolocek C, et al. Patterns of host genome-wide gene transcript abundance in the peripheral blood of patients with acute dengue hemorrhagic fever. J Infect Dis. 2007;195:1097-107.
- Labuda M, Randolph SE. Survival strategy of tick-borne encephalitis virus: cellular basis and environmental determinants. Zentralbl Bakteriol. 1999 Dec;289(5–7):513–24.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399-401.
- Barkhash AV, Babenko VN, Voevoda MI, Romaschenko AG. Association of IL28B and IL10 gene polymorphism with predisposition to tick-borne encephalitis in a Russian population. Ticks Tick Borne Dis. 2016;7:808-12.
- Barkhash AV, Perelygin AA, Babenko VN, Brinton MA, Voevoda MI. Single nucleotide polymorphism in the promoter region of the CD209 gene is associated with human predisposition to severe forms of tick-borne encephalitis. Antiviral Res. 2012;93:64-8.
- Kreil TR, Zimmermann K, Burger I, Attakpah E, Mannhalter JW, Eibl MM. Detection of tick-borne encephalitis virus by sample transfer, plaque assay and strand-specific reverse transcriptase polymerase chain reaction: what do we detect? J Virol Methods. 1997;68:1-8.
- Labuda M, Austyn JM, Zuffova E, et al. Importance of Localized Skin Infection in Tick-Borne Encephalitis Virus Transmission. 1996;219:357-66.
- Kaiser R. Tick-borne encephalitis. Infect Dis Clin North Am. 2008;22:561-75.
- Czupryna P, Moniuszko A, Pancewicz SA, Grygorczuk S, Kondrusik M, Zajkowska J. Tick-borne encephalitis in Poland in years 1993-2008 – epidemiology and clinical presentation. A retrospective study of 687 patients. Eur J Neurol. 2011;18:673-9.
- Hudopisk N, Korva M, Janet E, et al. Tick-borne encephalitis associated with consumption of raw goat milk, Slovenia, 2012. Emerg Infect Dis. 2013;19(5):806-8.
- Dumpis, U., Crook, D., Oksi, J., 1999. Tick-borne encephalitis. Infect. Dis. 1999;28(4):882–90.
- Holzmann H, Aberle SW, Stiasny K, et al. Tick-borne Encephalitis from eating Goat Cheese in a Mountain Region in Austria. Emerg Infect Dis. 2009;15(10):1671-3.
- Poponnikova T V, Subbotin A V. The clinical features of the acute period of mixed infection of tick-borne encephalitis and Ixodes tick-borne borreliosis in children. [Article in Russian]. Med Parazitol (Mosk). 2005:7-10.
- Poponnikova T V. Specific clinical and epidemiological features of tick-borne encephalitis in Western Siberia. Int J Med Microbiol. 2006;296:59-62.
- Mansfield KL, Johnson N, Phipps LP, Stephenson JR, Fooks AR, Solomon T. Tick-borne encephalitis virus – a review of an emerging zoonosis. J Gen Virol. 2009;90:1781-94.
- Kaiser R. Tick-borne encephalitis: Clinical findings and prognosis in adults. Wien Med Wochenschr. [Internet]. 2012;162:239-43.
- Haglund M, Günther G. Tick-borne encephalitis–pathogenesis, clinical course and long-term follow-up. 2003;21 Suppl 1:S11-8.
- Kaiser R. The clinical and epidemiological profile of tick-borne encephalitis in southern Germany 1994-98: a prospective study of 656 patients. Brain. 1999;122:2067–78.
- Růžek D, Dobler G, Mantke OD. Tick-borne encephalitis: Pathogenesis and clinical implications. Travel Med Infect Dis. 2010;8:223-32.
- Karelis G, Bormane A, Logina I, et al. Tick-borne encephalitis in Latvia 1973-2009: epidemiology, clinical features and sequelae. Eur J Neurol. 2012;19:62-8.
- Lotric-Furlan S, Avsic-Zupanc T, Strle F. An abortive form of tick-borne encephalitis (TBE)–a rare clinical manifestation of infection with TBE virus. Wien Klin Wochenschr. 2002;114:627-9.
- Prokopowicz D, Bobrowska E, Bobrowski M, Grzeszczuk A. Prevalence of antibodies against tick-borne encephalitis among residents of north-eastern Poland. Scand J Infect Dis. 1995;27:15-6.
- Gustafson R, Forsgren M, Gardulf A, Granström M, Svenungsson B. Clinical manifestations and antibody prevalence of Lyme borreliosis and tick-borne encephalitis in Sweden: a study in five endemic areas close to Stockholm. Scand J Infect Dis. 1993;25:595-603.
- Gritsun TS, Lashkevich VA, Gould EA. Tick-borne encephalitis. Antiviral Res. 2003;57:129-46.
- Hirano M, Yoshii K, Sakai M, Hasebe R, Ichii O, Kariwa H. Tick- borne flaviviruses alter membrane structure and replicate in dendrites of primary mouse neuronal cultures. J Gen Virol. 2014;95(Pt_4):849-61.
- Schnoor M, Parkos CA. Disassembly of endothelial and epithelial junctions during leukocyte transmigration. Front Biosci. 2008;13:6638-52.
- Zajkowska J, Moniuszko A, Czupryna P, et al. Chorea and Tick- Borne Encephalitis, Poland. Emerg Infect Dis. 2013;19:1544-5.
- Logar M, Bogovič P, Cerar D, Avšič-Županc T, Strle F. Tick-borne encephalitis in Slovenia from 2000 to 2004: Comparison of the course in adult and elderly patients. Wien Klin Wochenschr. 2006;118:702-7.
- Anić K, Soldo I, Perić L, Karner I, Barac B. Tick-borne encephalitis in eastern Croatia. Scand J Infect Dis. 1998;30:509-12.
- Kaiser R. [Epidemiology and progress of early summer meningoencephalitis in Baden-Württemberg between 1994 and 1999. A prospective study of 731 patients]. Dtsch Med Wochenschr. 2000;125:1147-53.
- Günther G, Haglund M, Lindquist L, Forsgren M, Sköldenberg B. Tick-bone encephalitis in Sweden in relation to aseptic meningoencephalitis of other etiology: a prospective study of clinical course and outcome. J Neurol. 1997;244:230-8.
- Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet. 2008;371:1861-71.
- Poponnikova T V. Specific clinical and epidemiological features of tick-borne encephalitis in Western Siberia. Int J Med Microbiol. 2006;296:59-62.
- Kaiser R, Holzmann H. Laboratory findings in tick-borne encephalitis–correlation with clinical outcome. Infection. 2000;28:78-84.
- Pichler A, Sellner J, Harutyunyan G, Sonnleitner A, Klobassa DS, Archelos-Garcia J-J, et al. Magnetic resonance imaging and clinical findings in adults with tick-borne encephalitis. J Neurol Sci. 2017;375:266-9.
- Lenhard T, Ott D, Jakob NJ, et al. Predictors, Neuroimaging Characteristics and Long-Term Outcome of Severe European Tick-Borne Encephalitis: A Prospective Cohort Study. Klein RS, editor. PLoS One. 2016;11:e0154143.
- Horger M, Beck R, Fenchel M, et al. Imaging findings in tick- borne encephalitis with differential diagnostic considerations. AJR Am J Roentgenol. 2012;199:420-7.
- von Stülpnagel C, Winkler P, Koch J, et al. MRI-imaging and clinical findings of eleven children with tick-borne encephalitis and review of the literature. Eur J Paediatr Neurol. 2016;20:45- 52.
- Beattie GC, Glaser CA, Sheriff H, et al. Encephalitis with thalamic and basal ganglia abnormalities: etiologies, neuroimaging, and potential role of respiratory viruses. Clin Infect Dis. 2013;56:825-32.
- Czupryna P, Tarasow E, Moniuszko-Malinowska A, et al. MRI and planimetric CT follow-up study of patients with severe tick-borne encephalitis. Infect Dis (Lond). 2016;48:74-81.
- Günther G, Haglund M, Mesko L, et al. Regional cerebral blood flow scintigraphy in tick-borne encephalitis and other aseptic meningoencephalitis. J Nucl Med. 1998;39:2055-61.
- Dietmann A, Putzer D, Beer R, et al. Cerebral glucose hypometabolism in Tick-Borne Encephalitis, a pilot study in 10 Patients. Int J Infect Dis. 2016;51:73-7.
- Lehtinen I, Halonen JP. EEG findings in tick-borne encephalitis. J Neurol Neurosurg Psychiatry. 1984;47:500-4.
- Juhasz C, Szirmai I. Spectral EEG Parameters in Patients with Tick-Borne Encephalitis: A Follow-Up Study. Clin EEG Neurosci. 1993;24:53-8.
- Haglund M, Forsgren M, Lindh G, Lindquist L. A 10-Year Follow-Up Study of Tick-Borne Encephalitis in the Stockholm Area and a Review of the Literature: Need for a Vaccination Strategy. Scand J Infect Dis. 1996;28:217-24.
- Kaiser R, Vollmer H, Schmidtke K, Rauer S, Berger W, Gores D. [Follow-up and prognosis of early summer meningoencephalitis]. 1997;68:324-30.
- Czupryna P, Moniuszko A, Pancewicz SA, Grygorczuk S, Kondrusik M, Zajkowska J. Tick-borne encephalitis in Poland in years 1993-2008 – epidemiology and clinical presentation. A retrospective study of 687 patients. Eur J Neurol. 2011;18:673-9.
- Kaiser R. Langzeitprognose bei primär myelitischer Manifestation der FSME. Nervenarzt [Internet]. 2011;82:1020- 5.
- Misić Majerus L, Daković Rode O, Ruzić Sabljić E. [Post- encephalitic syndrome in patients with tick-borne encephalitis]. Acta Med Croatica. 2009;63:269-78.
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol. 2005;64:506-12.
- Gelpi E, Preusser M, Laggner U, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. 2006;12:322-7.
- Lani R, Moghaddam E, Haghani A, Chang L-Y, AbuBakar S, Zandi K. Tick-borne viruses: A review from the perspective of therapeutic approaches. Ticks Tick Borne Dis. 2014;5:457-65.
- Ruzek D. Patogeneze klíšťové encefalitidy a možnosti antivirové terapie. Epidemiol Mikrobiol Imunol. 2015;64:204-9.
- Eyer L, Šmídková M, Nencka R, et al. Structure-activity relationships of nucleoside analogues for inhibition of tick- borne encephalitis virus. Antiviral Res. 2016;133:119-29.
- Elsterova J, Palus M, Sirmarova J, Kopecky J, Niller HH, Ruzek D. Tick-borne encephalitis virus neutralization by high dose intravenous immunoglobulin. Ticks Tick Borne Dis. 2017;8:253-8.