
Chapter 9:
Immunology of TBEV-Infection
Sara Gredmark-Russ and Renata Varnaite
Key Points
- Tick-borne encephalitis (TBE) is a viral infectious disease of the central nervous system caused by the tick-borne encephalitis virus (TBEV).
- TBE is usually a biphasic disease and in humans the virus can only be detected during the first (unspecific) phase of the disease.
- Pathogenesis of TBE is not well understood, but both direct viral effects and immune-mediated tissue damage of the central nervous system may contribute to the natural course of TBE.
- The effect of TBEV on the innate immune system has mainly been studied in vitro and in mouse models.
- Characterization of human immune responses to TBEV is primarily conducted in peripheral blood and cerebrospinal fluid, due to the inaccessibility of brain tissue for sample collection.
- Natural killer (NK) cells and T cells are activated during the second (meningo-encephalitic) phase of TBE. The potential involvement of other cell types has not been examined to date.
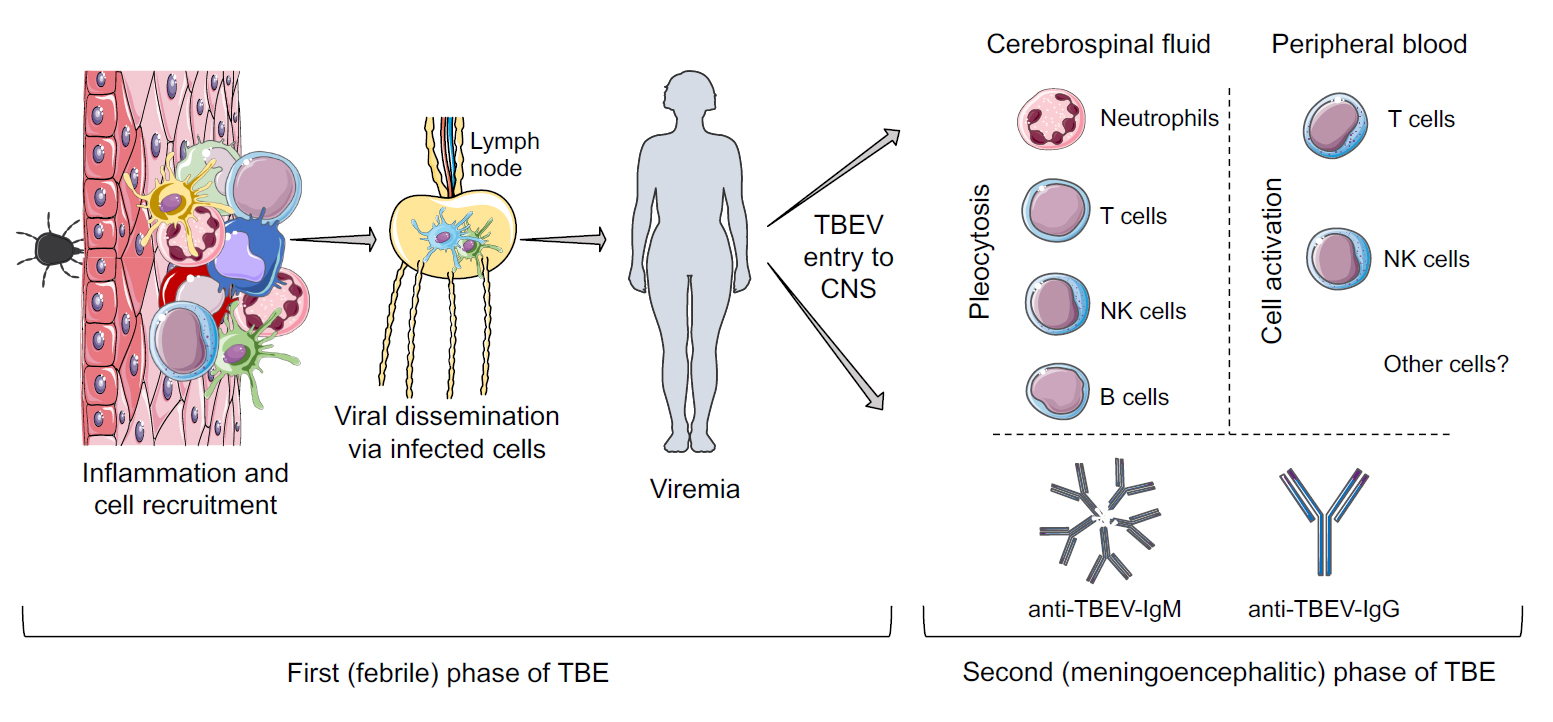
- Immune cells from peripheral blood, in particular neutrophils, T cells, B cells and NK cells, infiltrate into the cerebrospinal fluid of TBE patients.
Introduction
The immune system is a complex network of organs and processes within the host which protects from the invasion of pathogenic microorganisms. This network consists of an enormous variety of cells and molecules with specialized functions and is generally divided into innate and adaptive immunity. The innate immune system provides the first line of defense in infection and acts broadly against various pathogens, whereas the adaptive immune system generates a highly specialized response to individual pathogens. Adaptive immunity is also capable of “striking” harder upon secondary exposure to the same pathogen due to its ability to generate immunological memory. Importantly, the innate and adaptive immune systems function as allies to produce a more efficient total response than either one alone.
Tick-borne encephalitis (TBE) is a viral infectious disease of the central nervous system (CNS) caused by tick-borne encephalitis virus (TBEV). It is usually a biphasic disease manifesting with influenza-like febrile illness during the first (viremic/febrile) phase followed by a second (meningo-encephalitic) phase with neurological symptoms of different severity, ranging from meningitis to severe meningo-encephalitis (as reviewed in1). The first phase of TBE is challenging to study in humans, as infected individuals rarely seek medical attention. Therefore, sampling from blood or tissues from humans to study TBE is mostly possible during the second phase of disease. Interestingly, upon the emergence of neurological symptoms, the virus can no longer be detected in peripheral blood and cerebrospinal fluid (CSF).2 Whether as of this time the virus persists in other locations in the body (e.g. the brain parenchyma) has yet to be investigated.
Figure 1: TBE disease progression from TBEV-infected tick bite to the development of the second (meningoencephalitic) phase of TBE.
Click the image above to enlarge
The pathogenesis of TBE is also not completely understood. It may be attributed to either the direct viral cytolytic effects or the immune cell-mediated tissue damage, or both. TBEV viral proteins and immune cell infiltrates have been detected in neuronal tissues from fatal TBE cases supporting both mechanisms of pathogenesis – at least in such cases.3,4
In this chapter of the book, we aim to summarize the current understanding of the immune system responses to TBEV infection (Figure 1). First, we discuss the initial stages of TBE development including host barriers, viral spread, mechanisms of TBEV entry into the CNS and innate immune responses, most of which are delineated from in vitro or mouse models. We later review the adaptive immune system responses to TBEV infection, both humoral and cellular, from studies conducted primarily on human peripheral blood and CSF compartments.
Throughout this chapter, we also highlight the observed correlations between human immune responses and clinical TBE disease outcomes.
TBE disease progression
Barriers and local transmission of TBEV
Skin is one of the first physical barriers of the host that prevents the entry of pathogenic microorganisms. However, it is not purely a physical barrier, it is also equipped with many specialized immune cells, such as macrophages, mast cells, dendritic cell (DC) subsets, T cell subsets, and natural killer T cells ready to respond to any threatening microorganism.5 TBEV is mainly transferred to humans through the bite of infected ticks. Thus, the skin is the primary site of viral transmission from the tick’s saliva to the host. Virus transmission from the tick is facilitated by ‘‘saliva-activated transmission” factors within the tick’s saliva which contains components that interfere with the immune response, including factors that block and modulate inflammation, innate and acquired immunity, and wound healing.6 Already within the first hour of feeding, a stronger inflammatory microenvironment with increased cell recruitment is created at the TBEV-infected tick feeding site, as compared to uninfected tick feeding sites.7
After TBEV is transmitted, the cells residing in the skin tissue are exposed to the virus. Tick-feeding experiments in mice show that dendritic cells (DCs), mononuclear phagocytes and fibroblasts are the main cells to be infected by TBEV in the skin.7,8 Tick saliva further modulates TBEV infection of DCs by increasing their susceptibility to the virus and decreasing their ability to release inflammatory cytokines.9 Mononuclear phagocytes and DCs are believed to be involved in viral dissemination of TBEV, as these cells can migrate from the skin to the draining lymph nodes.
Viral dissemination and entry into the CNS
Systemic virus infection – viremia, is a common cause of febrile flu-like symptoms manifesting due to the immune response to a virus. It is therefore assumed that the first phase of TBE involving febrile symptoms is the result of immune responses to the systemic infection with TBEV. During this phase, TBEV RNA can be detected in human blood samples.10,11 As soon as anti-TBEV antibodies are detectable in the blood, viral RNA can usually no longer be found in blood or CSF samples.10,11 TBEV RNA has been detected in urine samples during the second (meningo-encephalitic) phase of the disease, and persistent viremia has been described in immunosuppressed patients.12,13
The exact route of TBEV entry into the CNS is unknown. TBEV antigen is found in brain tissue in autopsies from fatal cases of TBE, and the virus is selectively localized in the neurons.3 As for other, more well-studied neurotropic flaviviruses in this context, e.g. West-Nile virus (WNV) and Japanese Encephalitis virus (JEV), different ways of viral entry have been suggested, that may be dependent on blood-brain barrier (BBB) breakdown, passive diffusion of virus, or via infected-leukocytic trafficking.14 An additional mechanism that has been suggested is trans-neural invasion of virus into the CNS, via either peripheral somatic or the olfactory nerves.14 TBEV can infect various cells from the central nervous system in vitro, including brain microvascular endothelial cells in a BBB model.15-18 However, BBB breakdown does not seem to be a prerequisite for TBEV entry into the brain. In vitro studies show that the virus can cross the BBB via a transcellular pathway without altering the BBB integrity.18 Additionally, in a rodent TBE model BBB breakdown is primarily a result of cytokine release by the infected cells and BBB breakdown is not required for TBEV entry into the brain.19
Innate immune system and TBE
The innate immune system
The primary function of the innate immune system of the host is to prevent the entry of and colonization by pathogenic microorganisms, and if entry occurs, to limit the infection. All cells in the body, though to a varying extent, are “trained” to recognize and respond if such penetration occurs. The innate immune cell recognition of pathogens takes advantage of pattern recognition receptors (PRRs). PRRs detect pathogen-associated molecular patterns (PAMPs) to initiate protective immune responses and subsequent elimination of the “invaders”.20 Different classes of PRRs are involved in detection of viral infections, such as Toll-like Receptors (TLRs), cytoplasmic protein retinoic acid–inducible gene 1 (RIG-I) and structurally related melanoma differentiation-associated gene 5 (MDA5).21 Via different signalling pathways these molecules induce antiviral responses upon sensing viral PAMPs from different cellular compartments.21 Endosomal TLRs, for example, including TLR3, TLR7, TLR8 and TLR9, recognize viral nucleic acids, including double-stranded RNA, single-stranded RNA, and DNA.22 Upon ligand recognition, TLRs trigger the production of type I interferons (IFN) and inflammatory cytokines/chemokines to activate antiviral defense mechanisms and to initiate adaptive immune responses.22
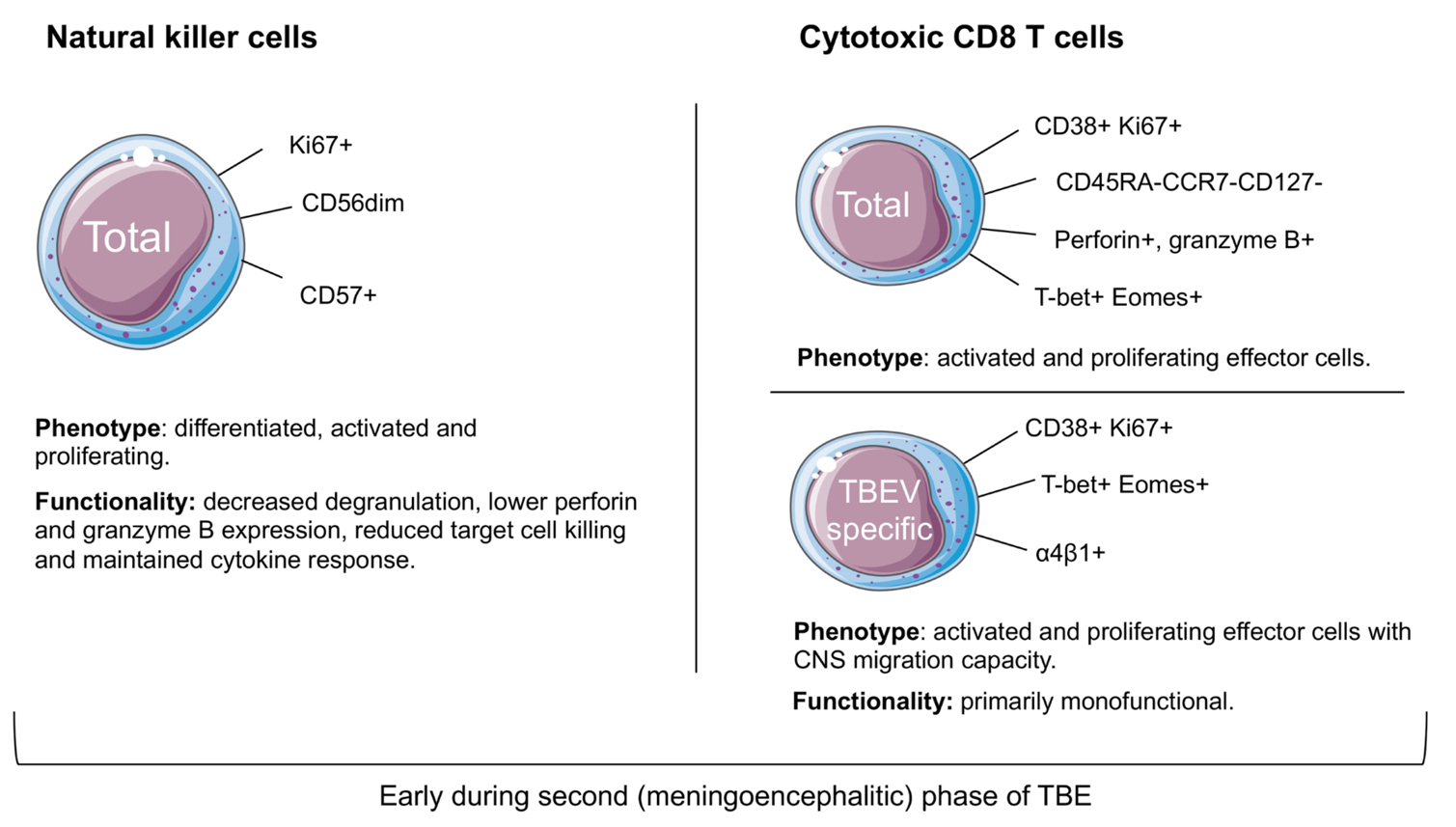
Figure 2: Cellular responses to human TBEV-infection in the peripheral blood compartment during the second phase of TBE.
Click the image above to enlarge
Type I IFN can be produced by most cell types and its receptors are widely distributed on the cell surface.23 Binding of IFNs produced by infected cells to the IFN receptor complex on the surrounding cells results in the expression of interferon-stimulated genes (ISG). ISGs have been shown to modulate/inhibit viral replication by inducing an antiviral state.24 In addition to interferons, cytokines and chemokines are also very important secreted factors of the immune response to pathogens.25 They orchestrate many processes during infection by controlling immune cell trafficking and determining the nature of the downstream immune responses. Important cells of the innate immune system are dendritic cells (DCs), phagocytes (neutrophils, monocytes and macrophages), cells releasing inflammatory mediators (basophils, mast cells and eosinophils) and the NK cells.
As the innate immune system is activated during the early stages of infection, it is difficult to study its role in TBEV-infection in humans. TBE patients are usually admitted to hospital very late during the infection, already after the adaptive immune system responses are initiated. Therefore, the majority of research on TBEV and the innate immunity are performed in mouse and in vitro models, also taking advantage of the natural attenuated Langat virus that belongs to the TBEV serocomplex.
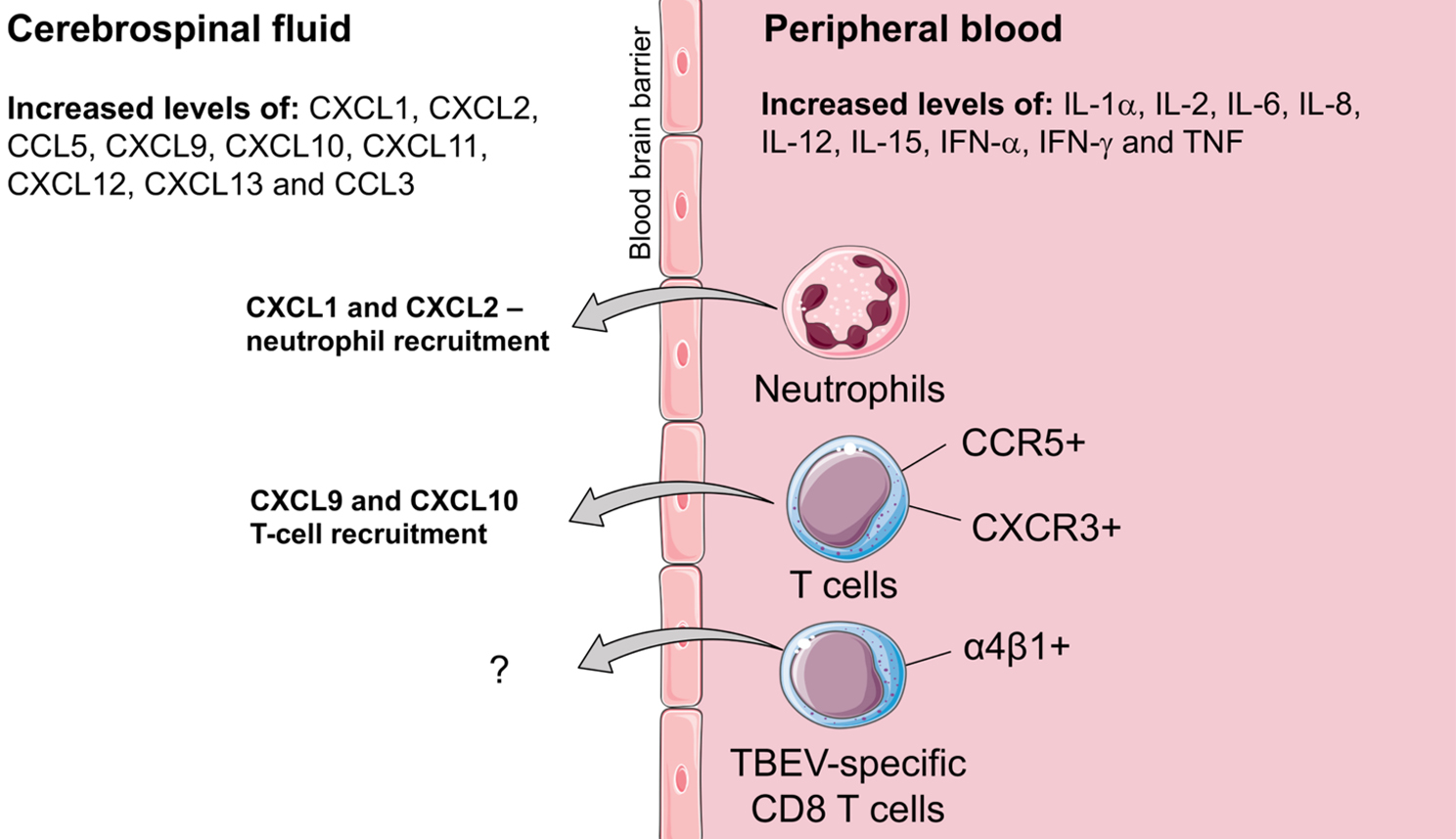
Figure 3: Expression of chemokines, cytokines and other signalling molecules in the cerebrospinal fluid (CSF) and peripheral blood, as well as their role in immune cells recruitment to the central nervous system in TBE.
Click the image above to enlarge
Pattern recognition receptor signalling and type I interferon response to TBEV
As for other viral infections, animal models and in vitro experiments demonstrate that type I IFN has a protective role against TBEV-infection. IFN-receptor-deficient mice infected with TBEV or Langat virus develop severe clinical symptoms and succumb to the infection, most likely due to unrestrained systemic viremia, and local inflammation induced by viral replication in the brain.26 Further experiments have suggested that interferon-beta promoter stimulator 1 (IPS-1), a downstream adaptor for MDA5 and RIG-1-like receptor signalling is important in controlling TBEV and Langat virus infection in mice.27 Knockout of IPS-1 leads to increased viral replication, release of inflammatory cytokines and immune cell infiltration in the CNS of Langat infected mice.
In vitro experiments demonstrate that astrocytes initiate a very early type I IFN antiviral response upon TBEV-infection thereby limiting viral replication and spread.28 RIG-1-like receptors are upregulated together with various ISGs in human neuronal derived cell lines by TBEV-infection.29 A number of ISGs have been shown to specifically target TBEV-infection. The Tripartite motif (TRIM) 79α protein restricts TBEV and Langat virus replication by mediating lysosome-dependent degradation of the NS5 protein.30 TRIM79α was also important for eliciting antiviral activity of IFNβ for inhibition of TBEV replication. Viperin (virus-inhibitory protein, endoplasmatic-associated, interferon inducible) has also been shown to restrict TBEV replication by proteasome-dependent de-gradation of NS3 viral protein, and reduced the stability of other TBEV proteins (prM, E, NS2A and NS2B) in the presence of NS3.31 Studies of polymorphisms in innate immune genes support the importance of innate immunity in TBE, as 5 different single nucleotide polymorphisms (SNPs) in the interferon-induced antiviral proteins oligo-adenylate synthetase 2 (OAS2) and 3 (OAS3) have been suggested to be associated with clinical TBE infection.32
Even though TLR signalling has been studied to some extent in other flavivirus infections33, the role of TLR in TBEV infection is not clear. A functional TLR3 receptor was suggested to be a risk factor for clinical TBE infection in adults, but not in children.34-36 TLR7 signalling has also been shown to have a role in controlling the replication of Langat virus as TLR7-deficiency in mice increases the virus burden in the CNS. However, increased viral burden does not seem to influence the level of neuropathogenesis.37 The mechanism for the increased viral replication in the neurons of these TLR7-deficient mice is not clear. However, lower levels of pro-inflammatory cytokines and chemokines are observed.
Innate response and its antagonism by TBEV
Many viruses induce activation of PRR and subsequent IFN signalling within hours of viral infection. Similarly, to other viruses, TBEV may also have many mechanisms to interfere with or evade the innate immunity. TBEV as a single-stranded RNA (ssRNA) virus produces double-stranded RNA (dsRNA) intermediates during replication. One of the earliest immune evasion strategies by TBEV is to hide its dsRNA from the cytoplasmic PRRs within the host cells by rearranging internal cell membranes.38 Inaccessibility of dsRNA for cytoplasmic PRRs delays the activation of interferon regulatory factor 3 (IRF-3), a key transcriptional regulator of type I IFN response. This results in a subsequent 24h delay of IFN production giving an opportunity for TBEV to replicate unhindered.38
Effective and early IFN responses are critical during viral infection, thus active antagonism of host proteins involved in IFN responses is another common viral mechanism of evasion. Viruses often use their own proteins to directly interact with and inhibit IFN signalling molecules. Studies on Langat virus have shown that viral nonstructural protein 5 (NS5) is an IFN antagonist and inhibits the JAK-STAT signal transduction pathway by blocking the phosphorylation of STAT1, STAT2, Tyk2 and Jak1.39 Similarly, TBEV NS5 protein was also found to block the phosphorylation of STAT1 by binding to the host membrane protein scribble (hScrib) resulting in inhibition of downstream IFN signalling.40 Another known target of TBEV NS5 is host protein prolidase (PEPD). Interaction of NS5 with PEPD is associated with decreased surface expression of type I IFN receptor subunit IFNAR1 resulting in reduced ISG expression.41 These findings highlight an important role of TBEV NS5 protein as a strong antagonist of type I IFN response.
Innate cellular responses in circulation during TBE
NK cells
Natural killer (NK) cells are cytotoxic innate lymphoid cells that are an important part of the immune response against viruses and tumor cells. NK cells represent a distinct population of lymphocytes that lack CD3 and express CD56. The two main NK cell populations in peripheral blood of healthy individuals are the cytotoxic CD56dimCD16+ NK cells, and the less cytotoxic CD56brightCD16– NK cells which produce larger amounts of cytokines upon activation.42 The ability of NK cells to distinguish between normal and infected cells is partly dependent on the surface MHC class I expression levels. In addition, NK cells express multiple activating and inhibitory receptors, and the state of NK activation or tolerance is dependent on a balance of the engagement of these receptors.43 NK cell cytotoxicity is mediated via three main pathways: 1) cell lysis of infected cells using perforin- and granzyme, 2) Fas ligand–mediated induction of apoptosis, 3) antibody-dependent cellular cytotoxicity, NK cells also produce cytokines and chemokines for communication with surrounding cells, thereby also bridging the innate and adaptive immune response.44
In patients suffering from TBE, NK cells are present in both peripheral blood and the CSF with higher percentages of NK cells residing in the blood.45 Even though the virus is not detected during the second phase of TBE, increased levels of cytokines that either activate or are produced by NK cells are detected in blood.46 In addition, NK cells are shown to be activated at early time points during the second phase of TBE (Figure 2). The TBEV-induced NK cell activation was predominantly seen in more differentiated NK cells (CD57+CD56dim). The activated NK cells had less expression of perforin, granzyme B, and Bcl-2, suggesting that the cells have already responded to target cells. In addition, CD56dim NK cells had a decreased responsiveness to target cells ex vivo but recovered their functional capacity during the convalescent phase of TBE. In contrast to the decreased response to target cells, the NK cells could respond to cytokine stimulation ex vivo throughout the infection. Interestingly, the characteristics of NK cell responses in TBE infection are different from those of other human viral infections. The release of cytotoxic granules early in NK cell activation may contribute to the pathogenesis in TBEV-infection.
Neutrophils
Neutrophils contribute to the inflammatory response and have phagocytic activity early during innate immune responses to viral infections. They are attracted to the bite site during tick feeding experiments and can also be infected by TBEV.8 Neutrophils are also present at high levels in human CSF early after TBE onset, slowly decreasing over time.47 However, despite decreasing numbers, neutrophil counts in CSF are higher during the convalescent phase in patients with persistent neurological symptoms.48 In TBE patients concentrations of chemokines signalling through CXCR1 and CXCR2 receptors are upregulated in the CSF suggesting a potential mechanism for neutrophil infiltration.48
In a mouse model using Langat virus, neutrophils have been suggested to mediate brain injury. Increased levels of neutrophils in the CNS were observed during late infection time points in CCR5 deficient mice, together with high levels of neutrophil attracting chemokines CXCL1 and CXCL2, higher viral load and increased apoptosis in the brain tissue. Depletion of neutrophils reversed this phenotype (Figure 3).49
Adaptive immune system and TBE
The adaptive immune system recognizes and selectively eliminates specific foreign microorganisms and toxic molecules, i.e. antigens. The adaptive immune system displays characteristic attributes including antigen specificity, immunologic memory, self-tolerance and non-self-discrimination. The key cells of adaptive immunity are the T and B lymphocytes, which express antigen-specific receptors on their cell surface. Adaptive immunity can be divided into humoral and cell-mediated.
Antigen presenting cells and antigen presentation during TBE
Dendritic cells (DCs) act as an important bridge between the innate and adaptive immune systems. DCs express many types of PRRs, thus enabling DCs to respond to various pathogens by the recognition of PAMPs.50 Antigen uptake and the engagement of PRRs induce processes of chemokine receptor switching, upregulation of co-stimulatory molecules and cytokine secretion. DCs become activated and mature after PRR stimulation resulting in their migration from tissues to lymph nodes where they can initiate T cell responses.
In order to specifically recognise a given antigen, naïve antigen-inexperienced T cells require antigen presentation in the form of a peptide by antigen-presenting cells (APC) via Histocompatibility Complex (MHC) molecules on their surface. APCs include DCs, monocytes/macrophages, Langerhans cells and B cells. DCs are the most potent antigen-presenting cells and are professional inducers of T cell responses. MHC-peptide complexes on DCs bind to the T-cell receptor (TCR) providing the first signal of activation. The secondary signal of co-stimulatory molecule engagement is required for full activation of T cells. The CD28 is a major co-stimulatory molecule on T cells, and it facilitates T cell activation upon binding to CD80 and CD86 on DCs.51 DCs are also producers of type I IFN that have multiple functions in adaptive immunity, such as T cell proliferation, CD8 T cell activation, B cell isotype switching and differentiation into plasma cells.50
Many flaviviruses, including Langat virus, can infect DCs in vitro.52 Infection of DCs results in impaired DC maturation and subsequently decreased T cell priming/proliferation.
However, when mouse dendritic cells were infected with 2 different strains of TBEV, DC maturation was instead induced, as measured by co-stimulatory molecule and MHC class II upregulation on the cell surface.53 Tick-feeding experiments on mice also showed that DCs and monocytes are locally infected by TBEV at the bite site, therefore potentially contributing to subsequent viral spread and the initiation of the adaptive immune responses.8
B lymphocytes and antibody responses during TBE
B cells carry a large variety of immunoglobulin (Ig) surface receptors that can directly recognize antigens and are responsible for specific antibody production. Naive B cells are activated after primary antigen encounter and initially produce antigen-specific IgM, and later IgG. Activated B cells later differentiate into plasma and memory B cells. Plasma cells are responsible for antibody secretion during the immune response, whereas memory B cells are responsible for the recall responses during repeated infection with the same pathogen.54
Many viral infections and vaccines give rise to long-lasting protective immunity consisting of pathogen-specific antibodies and memory B cells. TBEV infection also elicits an efficient B cell response. During the first (viremic) phase of TBE, anti-TBEV antibodies are generally not detected.11 However, anti-TBEV IgM- and IgG-antibodies appear in serum during the second phase of the disease.2 During the second phase of TBE, virus is rarely present in human serum, therefore detection of viral RNA using PCR is not optimal for TBE diagnosis. For this reason, diagnosis of TBE is primarily based on serology, i.e. presence of TBEV-specific IgM and IgG- antibodies in serum and CSF of patients.2
A number of studies have attempted to correlate humoral responses to TBE infection with clinical outcome. High anti-TBEV IgM antibody levels were detected early during TBE, decreasing over time in both serum and CSF, whereas IgG antibodies were detected later than IgM, peaked in the six-week convalescent samples and persisted for more than a year.55 The persistence of serum and CSF antibodies did not correlate to the disease severity, but patients with low levels of IgM antibodies in CSF during the early second phase of TBE, and patients with low TBEV-neutralizing antibody levels in serum suffered from a more severe disease.55,56 In addition, a more recent study found a higher concentration of anti-TBEV IgG antibodies in serum of patients with a milder disease as compared to those with a severe TBE.57 These studies may support a link between humoral immunity and TBE clinical outcome.
Mouse studies provide additional data that suggest that B cells contribute to the outcome of TBEV infection. Increase of CD19 mRNA levels in brain tissue of infected mice coincides with high levels of TBEV-neutralizing anti-bodies.58 These mice are also less susceptible to TBEV than mice producing low levels of neutralizing antibodies.58 However, the mice more susceptible to TBEV also exhibited strong cytokine/chemokine mRNA production in the brain, suggesting that other immuno-pathological mechanisms are involved in the disease outcome.
Even though the antibody response during TBE has been studied to some extent, the cellular aspects of B cell response, including phenotype and activation status, as well as the overall B cell role in TBE pathogenesis remain to be investigated.
T lymphocyte responses during TBE
T cells are characterized by the expression of the cell surface marker CD3, which forms a complex with the T cell specific receptors. Conventionally, the T cells are divided into two groups with different immune functions: T helper (CD4) and cytotoxic (CD8) cells, based on their surface expression of either CD4 or CD8 markers. Activated CD4 T cells secrete various cytokines that orchestrate the immune response by activating B cells, CD8 T cells, macrophages and other cells of the immune system. CD4 cells are restricted to recognizing peptides presented on MHC class II molecules on APCs, whereas the CD8 cells recognize peptides presented on MHC class I molecules. CD8 T cells have a cytolytic ability to kill infected host cells. The killing is mediated by the release of cytolytic proteins like perforin and granzyme. Most CD8 T cells are also efficient cytokine producers.
Adequate T cell responses, both CD4 and CD8, are important during viral infections. The effector CD8 T cells contribute to the clearance of the infection and provide long-lasting immunity. CD4 T cells have a central “helper” role to assist and activate B cells and CD8 T cells. After the naïve T cells encounter an antigen they differentiate into effector T cells, with the majority of the effector cells dying off after clearance of the infection, with only a small pool of cells remaining as memory cells.59 Memory cells can respond rapidly upon re-exposure to the same infectious agent.59 Different subsets of memory cells can be defined based on their phenotypic markers, with the central memory cells homing to secondary lymphoid organs and effector memory cells mostly found in non-lymphoid organs.59
T cell activation and phenotype during TBE
T cell activation and phenotype was investigated longitudinally in TBE patients during the second phase of the disease, from the time of hospitalization up to the convalescent period. Peripheral blood T cells were found to be activated (as determined by Ki67 and CD38 co-expression), with the activation peaking at one week after hospitalization (Figure 2).60 In contrast to CD8 T cells, CD4 T cells showed only low levels of activation at this time of infection.60 Activated CD8 T cells had increased expression of perforin and granzyme B and passed through an effector phase prior to differentiation into memory cells. TBEV-specific CD8 T cells were further shown to be mainly monofunctional in response to TBEV-peptide stimulation early after hospitalization, but became more polyfunctional in the convalescent phase.60 Additionally, TBEV-specific CD8 T cells express higher levels of α4- and β1-integrins than the bulk CD8 T cells, which may indicate their ability to migrate into the CNS.61
These data indicate that the primary CD8 T cell response to TBEV infection occurs during the second phase of TBE, as the peak of activation of CD8 T cells along with the occurrence of TBEV-specific CD8 T cells take place at about one week into the second phase of TBE.60,61 In a yellow fever vaccine-based infectious model the peak of CD8 T cell response was observed at day 15 after immunization.62 This may suggest a slight delay of CD8 T cell activation during TBEV infection as compared to the yellow fever vaccine-based infectious model. However, without access to patient samples during the first phase of TBE it is difficult to explain the exact kinetics of T cell responses.
Role of T cells in TBE pathogenesis
Even though T cells participate in the immune response to TBEV, the role of these cells in the outcome of TBE is not clear. One study suggests that T cell infiltration into the CNS during TBEV infection might contribute to a favourable disease outcome.49 In vivo studies of CCR5-deficient mice infected with Langat virus, show a delayed influx of CD4 and CD8 T cells into the CNS, increased viral replication and decreased survival of these mice, suggesting a protective role of T cells.49 Other conflicting studies suggest immunopathological rather than protective role of CD8 T cells in TBE. Brain tissue biopsies from fatal TBE cases show cytotoxic T cell infiltration in close proximity of TBEV-infected neurons.4 In addition, CD8 deficient mice have a prolonged survival as compared to immunocompetent mice during TBEV infection, and this effect is independent of viral load in the periphery or the brain.63 This immunopathology is primarily mediated by CD8 T cells and not CD4 T cells, as shown by shorter time of survival of immunodeficient SCID mice receiving CD8 T cells, whereas adoptive transfer of CD4 T cells increases the survival time.63 Yet, other mouse studies that compared mice challenged with TBEV that died or recovered, did not detect any differences in T cells numbers in the brains of the two groups, even if the cell numbers were increased in both groups as compared to uninfected mice.64 Interestingly, T cell receptor antigen specificity might determine the severity of TBEV-infection in mice, as T cell clones that express certain TCRs accumulate in the brains of mice dying from TBEV.65 These conflicting studies highlight the need for further research to understand the role of T cells in the context of TBE pathogenesis.
T cell antigen specificity in TBEV infection and vaccination
Antigen-specific T cells can be detected by artificially generated and fluorescently label-led peptide-MHC complexes.66,67 Antigen-specific T cells recognize the peptides present-ed on these complexes and bind to them. This binding can then be detected using flow cytometry. This is an invaluable tool to study virus-specific cells in patients including TBEV-infection.
In total, seven TBEV-specific peptides have been identified, and all of them are located in nonstructural (NS) proteins of the virus.60,61 The majority of previously identified CD8 T cell viral peptides in other flaviviral infections, such as YFV and DENV, are also derived from NS proteins.68-70
Immunodominant regions of viral proteins can be determined by stimulating cells with peptides based on the full viral protein sequences. Antigen specific cells upon binding to such peptides might initiate cytokine release (like IL-2) which can be measured for each peptide. Such studies on CD4 T cells after TBEV infection and vaccination identified immunodominant regions of structural viral proteins.71,72 Both vaccinated and infected individuals responded to similar regions of TBEV structural proteins, even if the response was higher in the vaccinated cohort. Another study showed that, full recombinant structural TBEV proteins trigger CD4 T cells, but not CD8 T cells in TBE-vaccinated individuals.73 Therefore, CD4 T cell responses seem to be skewed toward recognition of structural proteins, whereas CD8 T cell responses are skewed toward recognition of NS proteins.
Inflammation during TBE
Inflammation upon acute infections is an important part of the immune response essential for the elimination of pathogens. On the other hand, excessive inflammation may be harmful to the host. Many cells contribute to the inflammatory processes during infections to produce different cytokines, chemokines and growth factors. During TBEV infection there is both a systemic inflammatory response in the peripheral tissues, as well as a localised inflammation in the central nervous system (Figure 3). Numerous studies investigated the levels of cytokines and chemokines in serum and CSF of TBE patients, yet there are limited data on the role of inflammation in the pathogenesis of TBE.
Early during the second phase of TBE, significantly increased levels of cytokines, such as IL-1α, IL-2, IL-6, IL-8, IL-12, IL-15, IFN-α, IFN-γ and TNF can be detected in patient serum samples.46,74,75 The levels of these cytokines decline over time. Increased levels of growth factors, such as hepatocyte growth factor and vascular endothelial growth factor, as well as increased serum levels of matrix metallopeptidase-9 (MMP-9) have also been found in the sera during second phase of TBE.75,76 Increased levels of MMP-9 highlight the presence of local inflammation within the CNS in TBE patients, as increase of MMP-9 is associated with brain tissue damage.77 A polymorphism in MMP-9 gene (rs17576 SNP), which affects the function of this protein,78 was also found to predispose TBE patients with this SNP to develop CNS damage.79
Chemokines are a type of cytokines that mediate immune cell recruitment and activation in inflamed tissues. Chemokine gradient also determines the direction for the immune cell movement in and out of tissues.25 In the context of TBE patients, increased CXCL9 and CXCL10 levels in the CSF were shown to create a chemokine gradient between the CSF and serum, potentially resulting in the recruitment of CXCR3 receptor expressing T cells into the CNS.80-82 Even if a gradient between the CSF and serum could not be confirmed for CCL5 (RANTES), CXCL11, CXCL12, CXCL13, and CCL3, the concentration of these chemokines is also increased in the CSF of TBE patients.81,83,84 The level of CCL5 in CSF is correlated with pleocytosis, and activated CD4 T cells in CSF also expressed a high level of CCR5 (receptor for CCL5), further indicating that CCL5 acts as a chemoattractant to recruit cells into the CNS of TBE patients.84 The neutrophil chemoattractant CXCL1 and CXCL2 has also been shown to be increased in CSF early during TBE.48
In addition to chemokines, the levels of other cytokines in the CSF of TBE patients were assessed and correlated with clinical TBE outcome. A significantly increased concentration of IFN-γ, IL-4, IL-6 and IL-8 was found in the CSF of children who developed sequelae after TBE, as compared to the children who did not.85 In adults, low levels of IL-10 in the CSF later during the second phase of TBE (day 7-18) correlated with a more severe disease.86
CNS immune responses during TBE
The mechanisms underlying TBE pathogenesis in the CNS in humans are still largely unknown and under-explored. Relatively low mortality of TBE patients and inaccessibility of brain tissue samples are the main challenges in describing the immune mechanisms taking place in the CNS in humans. Therefore, most of the research on immune cell subsets within the CNS is performed on CSF, a fluid that is separated from peripheral circulation via the BBB and is in direct contact with the brain and spinal cord. Even though the cellular constitution of CSF reflects which cell types selectively migrate through the BBB from peripheral blood during CNS infections, it does not necessarily fully represent the composition of the immune cells within infected brain tissue. However, selective migration of certain cell types may contribute to defining the mechanisms for pathogen clearance and immunopathogenesis and may also predict TBE disease outcome.
Pathogenesis in the CNS during TBEV infection may be attributed to direct viral effect and immune-mediated tissue damage, both of which are supported by the detection of TBEV viral proteins and immune cell infiltrates in neuronal tissues from cases of fatal TBE.3,4 The mechanism for virus passage through the BBB into the brain is not yet defined, as discussed more in detail under the section “Viral dissemination and entry into the CNS”. Neurons are believed to be the primary targets for TBEV in the CNS,3,87 but other brain cells are also infected in vitro.15-17
Immune cell infiltration into the CSF, defined as pleocytosis, is a common event during CNS infections. Early during the second (meningoencephalitic) phase of TBE, CSF contains a higher proportion of neutrophils, whereas mononuclear cells steadily increase overtime to become the dominant cell type.47 Importantly, immune cells such as CD4 and CD8 T cells, NK cells and B cells are also present in the CSF of TBE patients, with T cell frequencies being higher than in blood, indicating selective migration of these cells through the BBB.45 Most previous studies on CNS infiltration of T cells, however, were performed in human neuroinflammatory diseases and in animal models for auto-immune and viral infections, including HIV.88-92
In general, virus-specific effector T cells are recruited to the CNS during infections by chemokines and integrins.88-90,92 To date, the exact mechanism for the recruitment of T cells (including TBEV-specific T cells) into the CNS during TBE is not clear, however certain chemokines and chemokine receptors were suggested to be involved. For example, infiltrating CCR5 and CXCR3-expressing T cells seem to have a role in TBE in humans (Figure 3). Chemokine CXCL10 (ligand for CXCR3) and CCL5 (ligand for CCR5) levels in the CSF of TBE patients are increased together with higher CCR5 expression on infiltrated CD4 T cells as compared to blood.80,81,84 Interestingly, a mutation in CCR5 is associated with a more severe course of the disease.36,9
In mouse models, TBEV infection induces CCL5 expression accompanied by increased immune cell infiltration into the CNS.94 Blocking of CCL5 reduced cell infiltration and extended the survival of mice after TBEV infection. In vitro TBEV infection of human glioblastoma cell lines and primary astrocytes by TBEV demonstrated that increased CCL5 expression is mediated by the viral TBEV protein NS5.94,95
An integrin role in T cell CNS recruitment during TBE is discussed in a recent study on TBEV-specific CD8 T cell and their expression of α4-integrin and β1-integrin.61 Almost all of the TBEV-specific CD8 T cells from peripheral blood express α4-integrin and β1-integrin early during second phase of TBE (1 and 3 weeks), whereas the bulk CD8 T cells expressed lower levels of integrins. Expression of α4β1 is associated with the ability to infiltrate tissues and cross the BBB.88-91 The same study, however, did not detect higher CXCR3 expression on TBEV-specific CD8 T cells as compared to bulk CD8 T cells. This may be due to the majority of TBEV-specific CD8 T cells residing in the CSF during patient sampling or by the possibility that CXCR3 is not crucial for CD8 T cell migration across the BBB in TBEV-infection. Further investigations on the mechanism for T cell migration into the CNS during TBE are required in order to explain the local CNS pathogenesis of this disease.
Host factors and TBE disease
As for most human infections, the clinical outcome of TBE is extremely variable, ranging from asymptomatic to lethal. A more severe TBE is associated with increased age, severity of symptoms during the first (febrile) phase, low neutralizing antibody titers at onset and low early CSF IgM response (as reviewed in1). The risk of developing TBE after exposure to the virus may also vary between individuals. For instance, an epidemiological study in Sweden measured seroprevalence for TBEV in an endemic area and found that only 25% of individuals who were seropositive for TBEV developed clinical TBE, suggesting that only 25% of naturally infected persons may develop disease, while 75% of the infections are non-symptomatic.96
Clinical appearance and the progression of TBE may also be related to host genetic factors. Studies on TBE in this context have thus far not been able to correlate susceptibility to TBE or disease severity to one single host genetic factor, but a few candidates have been suggested including CCR5Δ32 polymorphism,93 a functional TLR3 receptor,34-36 5 different SNPs in the interferon-induced antiviral proteins oligoadenylate synthetase 2 (OAS2) and 3 (OAS3),32 2 SNPs in the promoter region of CD209 (encoding dendritic cell-specific intercellular adhesion molecule (ICAM)-3 grabbing non-integrin (DC-SIGN)) expressed on the surface of dendritic cells,97 and SNPs in interleukin 28B (IL28B) and interleukin 10 (IL10).98 In a more recent study, the rs17576 SNP in the MMP-9 gene predisposed TBE patients for CNS damage.79
Conclusions
TBE is a complex and rather understudied disease in the context of human immune system responses. In vitro experiments, animal models, as well as research in humans have greatly contributed to describing TBEV infection and defining the mechanism of TBE disease progression, however, many aspects of it remain to be investigated further.
It is clear that TBEV is a potent inducer of innate immunity, but at the same time the virus is capable of antagonizing certain pathways of innate immune responses. Adaptive immune system responses are also initiated during TBE as reflected by anti-TBEV antibody presence in serum, as well as NK and T cell activation in peripheral blood of TBE patients. Local pathogenesis in the central nervous system in TBE may be attributed to both direct viral effects and immune mediated tissue damage, but the exact mechanism is unclear. More research is needed in order to fully understand the development of TBE in order to create effective and specific therapeutic strategies.
Acknowledgments
SGR is supported by the Marianne and Marcus Wallenberg Foundation. We sincerely thank Servier Medical Art (http://smart.servier.com/) for providing high-quality graphics, which we modified and compiled to create the figures for this chapter of the book.
Contact:
sara.gredmark.russ@ki.se
Citation:
Gredmark-Russ S, Varnaite R. Immunology of TBEV infection. Chapter 9. In: Dobler G, Erber W, Bröker M, Schmitt HJ, eds. The TBE Book. 6th ed. Singapore: Global Health Press;2023. doi:10.33442/26613980_9-6
References
- Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet. 2008;371:1861-71.
- Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine. 2003;21 Suppl 1:S36-40.
- Gelpi E, Preusser M, Garzuly F, Holzmann H, Heinz FX, Budka H. Visualization of Central European tick-borne encephalitis infection in fatal human cases. J Neuropathol Exp Neurol. 2005;64:506-12.
- Gelpi E, Preusser M, Laggner U, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. 2006;12:322-7.
- Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679-91.
- Kazimirova M, Thangamani S, Bartikova P, et al. Tick-Borne Viruses and Biological Processes at the Tick-Host-Virus Interface. Front Cell Infect Microbiol. 2017;7:339.
- Thangamani S, Hermance ME, Santos RI, et al. Transcriptional Immunoprofiling at the Tick-Virus-Host Interface during Early Stages of Tick-Borne Encephalitis Virus Transmission. Front Cell Infect Microbiol. 2017;7:494.
- Labuda M, Austyn JM, Zuffova E, et al. Importance of localized skin infection in tick-borne encephalitis virus transmission. Virology. 1996;219:357-66.
- Fialova A, Cimburek Z, Iezzi G, Kopecky J. Ixodes ricinus tick saliva modulates tick-borne encephalitis virus infection of dendritic cells. Microbes Infect. 2010;12:580-5.
- Saksida A, Duh D, Lotric-Furlan S, Strle F, Petrovec M, Avsic-Zupanc T. The importance of tick-borne encephalitis virus RNA detection for early differential diagnosis of tick-borne encephalitis. J Clin Virol. 2005;33:331-5.
- Saksida A, Jakopin N, Jelovsek M, et al. Virus RNA Load in Patients with Tick-Borne Encephalitis, Slovenia. Emerg Infect Dis. 2018;24:1315-23.
- Veje M, Studahl M, Norberg P, et al. Detection of tick-borne encephalitis virus RNA in urine. J Clin Microbiol. 2014;52:4111-2.
- Caracciolo I, Bassetti M, Paladini G, et al. Persistent viremia and urine shedding of tick-borne encephalitis virus in an infected immunosuppressed patient from a new epidemic cluster in North-Eastern Italy. J Clin Virol. 2015;69:48-51.
- Suen WW, Prow NA, Hall RA, Bielefeldt-Ohmann H. Mechanism of West Nile virus neuroinvasion: a critical appraisal. Viruses. 2014;6:2796-825.
- Ruzek D, Vancova M, Tesarova M, Ahantarig A, Kopecky J, Grubhoffer L. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J Gen Virol. 2009;90:1649-58.
- Palus M, Bily T, Elsterova J, et al. Infection and injury of human astrocytes by tick-borne encephalitis virus. J Gen Virol. 2014;95:2411-26.
- Bily T, Palus M, Eyer L, Elsterova J, Vancova M, Ruzek D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci Rep. 2015;5:10745.
- Palus M, Vancova M, Sirmarova J, Elsterova J, Perner J, Ruzek D. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology. 2017;507:110-22.
- Ruzek D, Salat J, Singh SK, Kopecky J. Breakdown of the blood-brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T-cells. PLoS One. 2011;6:e20472.
- Mogensen TH. Pathogen recognition and inflammatory signalling in innate immune defenses. Clin Microbiol Rev. 2009;22:240-73.
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131-7.
- Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305-15.
- de Weerd NA, Nguyen T. The interferons and their receptors–distribution and regulation. Immunol Cell Biol. 2012;90:483-91.
- Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519-25.
- Borish LC, Steinke JW. 2. Cytokines and chemokines. J Allergy Clin Immunol. 2003;111:S460-75.
- Weber E, Finsterbusch K, Lindquist R, et al. Type I interferon protects mice from fatal neurotropic infection with Langat virus by systemic and local antiviral responses. J Virol. 2014;88:12202-12.
- Kurhade C, Zegenhagen L, Weber E, et al. Type I Interferon response in olfactory bulb, the site of tick-borne flavivirus accumulation, is primarily regulated by IPS-1. J Neuroinflammation. 2016;13:22.
- Lindqvist R, Mundt F, Gilthorpe JD, et al. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation. 2016;13:277.
- Selinger M, Wilkie GS, Tong L, et al. Analysis of tick-borne encephalitis virus-induced host responses in human cells of neuronal origin and interferon-mediated protection. J Gen Virol. 2017;98:2043-60.
- Taylor RT, Lubick KJ, Robertson SJ, et al. TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe. 2011;10:185-96.
- Panayiotou C, Lindqvist R, Kurhade C, et al. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J Virol. 2018;92.
- Barkhash AV, Perelygin AA, Babenko VN, et al. Variability in the 2′-5′-oligoadenylate synthetase gene cluster is associated with human predisposition to tick-borne encephalitis virus-induced disease. J Infect Dis. 2010;202:1813-8.
- Guo HY, Zhang XC, Jia RY. Toll-Like Receptors and RIG-I-Like Receptors Play Important Roles in Resisting Flavivirus. J Immunol Res. 2018;2018:6106582.
- Kindberg E, Vene S, Mickiene A, Lundkvist A, Lindquist L, Svensson L. A functional Toll-like receptor 3 gene (TLR3) may be a risk factor for tick-borne encephalitis virus (TBEV) infection. J Infect Dis. 2011;203:523-8.
- Barkhash AV, Voevoda MI, Romaschenko AG. Association of single nucleotide polymorphism rs3775291 in the coding region of the TLR3 gene with predisposition to tick-borne encephalitis in a Russian population. Antiviral Res. 2013;99:136-8.
- Mickiene A, Pakalniene J, Nordgren J, et al. Polymorphisms in chemokine receptor 5 and Toll-like receptor 3 genes are risk factors for clinical tick-borne encephalitis in the Lithuanian population. PLoS One. 2014;9:e106798.
- Baker DG, Woods TA, Butchi NB, et al. Toll-like receptor 7 suppresses virus replication in neurons but does not affect viral pathogenesis in a mouse model of Langat virus infection. J Gen Virol. 2013;94:336-47.
- Overby AK, Popov VL, Niedrig M, Weber F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J Virol. 2010;84:8470-83.
- Best SM, Morris KL, Shannon JG, et al. Inhibition of interferon-stimulated JAK-STAT signalling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J Virol. 2005;79:12828-39.
- Werme K, Wigerius M, Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell Microbiol. 2008;10:696-712.
- Lubick KJ, Robertson SJ, McNally KL, et al. Flavivirus Antagonism of Type I Interferon Signalling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe. 2015;18:61-74.
- Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633-40.
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495-502.
- Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163-94.
- Tomazic J, Ihan A. Flow cytometric analysis of lymphocytes in cerebrospinal fluid in patients with tick-borne encephalitis. Acta Neurol Scand. 1997;95:29-33.
- Blom K, Braun M, Pakalniene J, et al. NK Cell Responses to Human Tick-Borne Encephalitis Virus Infection. J Immunol. 2016;197:2762-71.
- Jeren T, Vince A. Cytologic and immunoenzymatic findings in CSF from patients with tick-borne encephalitis. Acta Cytol. 1998;42:330-4.
- Grygorczuk S, Swierzbinska R, Kondrusik M, et al. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J Neuroinflammation. 2018;15:115.
- Michlmayr D, Bardina SV, Rodriguez CA, Pletnev AG, Lim JK. Dual Function of Ccr5 during Langat Virus Encephalitis: Reduction in Neutrophil-Mediated Central Nervous System Inflammation and Increase in T Cell-Mediated Viral Clearance. J Immunol. 2016;196:4622-31.
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987-95.
- Van Gool SW, Vandenberghe P, de Boer M, Ceuppens JL. CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol Rev. 1996;153:47-83.
- Robertson SJ, Lubick KJ, Freedman BA, Carmody AB, Best SM. Tick-borne flaviviruses antagonize both IRF-1 and type I IFN signalling to inhibit dendritic cell function. J Immunol. 2014;192:2744-55.
- Shevtsova AS, Motuzova OV, Kuragina VM, et al. Lethal Experimental Tick-Borne Encephalitis Infection: Influence of Two Strains with Similar Virulence on the Immune Response. Front Microbiol. 2016;7:2172.
- Dorner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27:384-92.
- Gunther G, Haglund M, Lindquist L, Skoldenberg B, Forsgren M. Intrathecal IgM, IgA and IgG antibody response in tick-borne encephalitis. Long-term follow-up related to clinical course and outcome. Clin Diagn Virol. 1997;8:17-29.
- Kaiser R, Holzmann H. Laboratory findings in tick-borne encephalitis-correlation with clinical outcome. Infection. 2000;28:78-84.
- Bogovic P, Lotric-Furlan S, Avsic-Zupanc T, Lusa L, Strle F. Factors associated with severity of tick-borne encephalitis: A prospective observational study. Travel Med Infect Dis. 2018; 26:25-31.
- Palus M, Vojtiskova J, Salat J, et al. Mice with different susceptibility to tick-borne encephalitis virus infection show selective neutralizing antibody response and inflammatory reaction in the central nervous system. J Neuroinflammation. 2013;10:77.
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749-61.
- Blom K, Braun M, Pakalniene J, et al. Specificity and dynamics of effector and memory CD8 T cell responses in human tick-borne encephalitis virus infection. PLoS Pathog. 2015;11:e1004622.
- Lampen M, Uchtenhagen H, Blom K, et al. Breadth and Dynamics of HLA-A2- and HLA–B7-restricted CD8+ T Cell Responses against Non-structural Viral Proteins in Acute Human Tick-borne Encephalitis Virus Infection. ImmunoHorizons. 2018;2:172-84.
- Blom K, Braun M, Ivarsson MA, et al. Temporal dynamics of the primary human T cell response to yellow fever virus 17D as it matures from an effector- to a memory-type response. J Immunol. 2013;190:2150-8.
- Ruzek D, Salat J, Palus M, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. 2009;384:1-6.
- Hayasaka D, Nagata N, Fujii Y, et al. Mortality following peripheral infection with tick-borne encephalitis virus results from a combination of central nervous system pathology, systemic inflammatory and stress responses. 2009;390:139-50.
- Fujii Y, Hayasaka D, Kitaura K, Takasaki T, Suzuki R, Kurane I. T-cell clones expressing different T-cell receptors accumulate in the brains of dying and surviving mice after peripheral infection with far eastern strain of tick-borne encephalitis virus. Viral Immunol. 2011;24:291-302.
- Rodenko B, Toebes M, Hadrup SR, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1:1120-32.
- Andersen RS, Kvistborg P, Frosig TM, et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat Protoc. 2012;7:891-902.
- Akondy RS, Monson ND, Miller JD, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183:7919-30.
- Miller JD, van der Most RG, Akondy RS, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710-22.
- Weiskopf D, Angelo MA, de Azeredo EL, et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc Natl Acad Sci U S A. 2013;110:E2046-53.
- Schwaiger J, Aberle JH, Stiasny K, et al. Specificities of human CD4+ T cell responses to an inactivated flavivirus vaccine and infection: correlation with structure and epitope prediction. J Virol. 2014;88:7828-42.
- Aberle JH, Schwaiger J, Aberle SW, et al. Human CD4+ T Helper Cell Responses after Tick-Borne Encephalitis Vaccination and Infection. PLoS One. 2015;10:e0140545.
- Gomez I, Marx F, Saurwein-Teissl M, Gould EA, Grubeck-Loebenstein B. Characterization of tick-borne encephalitis virus-specific human T lymphocyte responses by stimulation with structural TBEV proteins expressed in a recombinant baculovirus. Viral 2003;16:407-14.
- Atrasheuskaya AV, Fredeking TM, Ignatyev GM. Changes in immune parameters and their correction in human cases of tick-borne encephalitis. Clin Exp Immunol. 2003;131:148-54.
- Palus M, Formanova P, Salat J, Zampachova E, Elsterova J, Ruzek D. Analysis of serum levels of cytokines, chemokines, growth factors, and monoamine neurotransmitters in patients with tick-borne encephalitis: identification of novel inflammatory markers with implications for pathogenesis. J Med Virol. 2015;87:885-92.
- Palus M, Zampachova E, Elsterova J, Ruzek D. Serum matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 levels in patients with tick-borne encephalitis. J Infect. 2014;68:165-9.
- Kang X, Li Y, Wei J, et al. Elevation of matrix metalloproteinase-9 level in cerebrospinal fluid of tick-borne encephalitis patients is associated with IgG extravassation and disease severity. PLoS One. 2013;8:e77427.
- Zhang B, Henney A, Eriksson P, Hamsten A, Watkins H, Ye S. Genetic variation at the matrix metalloproteinase-9 locus on chromosome 20q12.2-13.1. Hum Genet. 1999;105:418-23.
- Barkhash AV, Yurchenko AA, Yudin NS, et al. A matrix metalloproteinase 9 (MMP9) gene single nucleotide polymorphism is associated with predisposition to tick-borne encephalitis virus-induced severe central nervous system disease. Ticks Tick Borne Dis. 2018;9:763-7.
- Lepej SZ, Misic-Majerus L, Jeren T, et al. Chemokines CXCL10 and CXCL11 in the cerebrospinal fluid of patients with tick-borne encephalitis. Acta Neurol Scand. 2007;115:109-14.
- Zajkowska J, Moniuszko-Malinowska A, Pancewicz SA, et al. Evaluation of CXCL10, CXCL11, CXCL12 and CXCL13 chemokines in serum and cerebrospinal fluid in patients with tick-borne encephalitis (TBE). Adv Med Sci. 2011;56:311-7.
- Koper OM, Kaminska J, Grygorczuk S, Zajkowska J, Kemona H. CXCL9 concentrations in cerebrospinal fluid and serum of patients with tick-borne encephalitis. Arch Med Sci. 2018;14:313-20.
- Grygorczuk S, Zajkowska J, Swierzbinska R, Pancewicz S, Kondrusik M, Hermanowska-Szpakowicz T. Elevated concentration of the chemokine CCL3 (MIP-1alpha) in cerebrospinal fluid and serum of patients with tick-borne encephalitis. Adv Med Sci. 2006;51:340-4.
- Grygorczuk S, Osada J, Parczewski M, et al. The expression of the chemokine receptor CCR5 in tick-borne encephalitis. J Neuroinflammation. 2016;13:45.
- Fowler A, Ygberg S, Bogdanovic G, Wickstrom R. Biomarkers in Cerebrospinal Fluid of Children With Tick-borne Encephalitis: Association With Long-term Outcome. Pediatr Infect Dis J. 2016;35:961-6.
- Gunther G, Haglund M, Lindquist L, et al. Tick-borne encephalitis is associated with low levels of interleukin-10 in cerebrospinal fluid. Infect Ecol Epidemiol. 2011;1.
- Hirano M, Yoshii K, Sakai M, Hasebe R, Ichii O, Kariwa H. Tick-borne flaviviruses alter membrane structure and replicate in dendrites of primary mouse neuronal cultures. J Gen Virol. 2014;95:849-61.
- Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17:179-94.
- Ifergan I, Kebir H, Alvarez JI, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain. 2011;134:3560-77.
- Martin-Blondel G, Pignolet B, Tietz S, et al. Migration of encephalitogenic CD8 T cells into the central nervous system is dependent on the alpha4beta1-integrin. Eur J Immunol. 2015;45:3302-12.
- Rothhammer V, Muschaweckh A, Gasteiger G, et al. alpha4-integrins control viral meningoencephalitis through differential recruitment of T helper cell subsets. Acta Neuropathol. Commun 2014;2:27.
- Shacklett BL, Cox CA, Wilkens DT, et al. Increased adhesion molecule and chemokine receptor expression on CD8+ T cells trafficking to cerebrospinal fluid in HIV-1 infection. J Infect Dis. 2004;189:2202-12.
- Kindberg E, Mickiene A, Ax C, et al. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tick-borne encephalitis. J Infect Dis. 2008;197:266-9.
- Zheng Z, Yang J, Jiang X, et al. Tick-Borne Encephalitis Virus Nonstructural Protein NS5 Induces RANTES Expression Dependent on the RNA-Dependent RNA Polymerase Activity. J Immunol. 2018;201:53-68.
- Zhang X, Zheng Z, Liu X, et al. Tick-borne encephalitis virus induces chemokine RANTES expression via activation of IRF-3 pathway. J Neuroinflammation. 2016;13:209.
- Gustafson R, Svenungsson B, Gardulf A, Stiernstedt G, Forsgren M. Prevalence of tick-borne encephalitis and Lyme borreliosis in a defined Swedish population. Scand J Infect Dis. 1990;22:297-306.
- Barkhash AV, Perelygin AA, Babenko VN, Brinton MA, Voevoda MI. Single nucleotide polymorphism in the promoter region of the CD209 gene is associated with human predisposition to severe forms of tick-borne encephalitis. Antiviral Res. 2012;93:64-8.
- Barkhash AV, Babenko VN, Voevoda MI, Romaschenko AG. Association of IL28B and IL10 gene polymorphism with predisposition to tick-borne encephalitis in a Russian population. Ticks Tick Borne Dis. 2016;7:808-12.